Nonacog gamma (▼Rixubis®, Baxalta) en hemofilia B
 Nº399
Nº399

Resumen
El nonacog gamma es una forma recombinante del Factor IX de coagulación. Ha sido autorizada para el tratamiento y profilaxis de hemorragias en pacientes con hemofilia B (déficit de Factor IX). Los datos acumulados de los tres estudios clínicos disponibles han demostrado la equivalencia farmacocinética del nonacog gamma con respecto al factor IX de origen extractivo; asimismo, se han establecido adecuadamente su eficacia y su seguridad, tanto en uso profiláctico como a demanda, incluyendo pacientes pediátricos y sometidos a intervenciones quirúrgicas. Aparentemente, no hay diferencias entre el nonacog gamma y los medicamentos con factor IX de origen extractivo disponibles actualmente, ya que a estas alturas las exigencias de seguridad y calidad de estos últimos son lo suficientemente elevadas como para que tal origen condicione su uso clínico.
ASPECTOS FISIOPATOLÓGICOS
La coagulación es un proceso extremadamente complejo que incluye una serie de reacciones generadas en la superficie de las células, que expresan en su superficie el factor tisular (FT), y cuyo objetivo es la formación de trombina en sitios de lesión vascular. La denominada de cascada de la coagulación considera un proceso de 3 fases: inicio, amplificación y acción de la trombina. La fase de inicio se produce tras la lesión vascular, cuando el factor VII (proconvertina) se une a las células que presentan en su superficie el factor tisular de tromboplastina (factor III), lo que conduce a la liberación proteolítica de la convertina (VIIa).
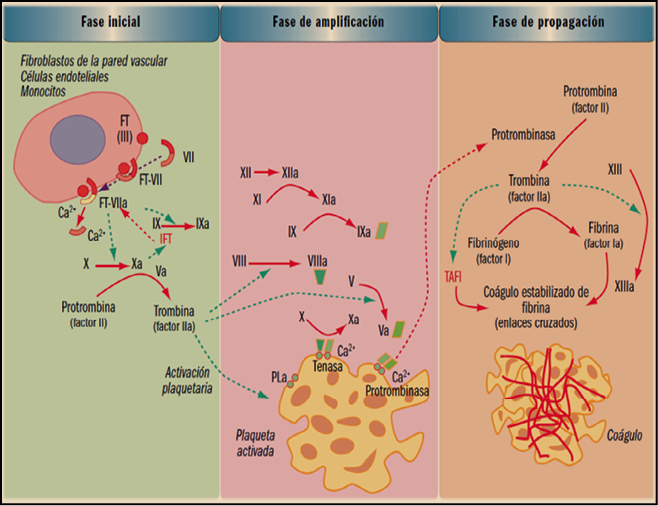
El complejo FT-VIIa facilita la generación de los factores Xa y IXa y esto causa la producción de una pequeña cantidad de trombina que inicia el proceso. La fase de amplificación se centra en la superficie de las plaquetas, activadas por la trombina, que acumulan en su superficie factores y cofactores y permiten su ensamblaje, dando lugar a complejos con actividad enzimática, como tenasa o protrombinasa, que facilitaran la fase de propagación, con la formación de grandes cantidades de trombina que favorecen la generación de fibrina y su polimerización, para dar lugar al coagulo estable, según se indica en la figura 1.
Los trastornos asociados a una deficiencia congénita en la producción de determinados factores de la coagulación sanguínea constituyen uno de los ejemplos típicos de las metabolopatías congénitas de mayor impacto. Aunque se trata de patologías poco frecuentes, su incidencia en varias de las familias reales europeas – debido a la estrecha endogamia – las popularizó.
La más común de las coagulopatías hereditarias – en realidad, la menos infrecuente – es la enfermedad de von Willebrand, un trastorno congénito transmitido autosómicamente, caracterizado por el déficit cualitativo y/o cuantitativo del factor de von Willebrand (Factor vW). Se ha calculado que en determinadas áreas puede llegar a afectar al 1% de la población, aunque los pacientes con problemas hemorrágicos graves no son numerosos. El déficit de factor VIII se conoce como hemofilia A y afecta 1 de cada 10.000 personas a nivel mundial; por tanto, se puede considerar como una enfermedad rara (menos de 5 casos por 10.000 habitantes, en la Unión Europea), al igual que el resto de la coagulopatías hereditarias, aún menos frecuentes que la hemofilia A, salvo la mencionada enfermedad de von Willebrand. Por su parte, la deficiencia de factor IX se conoce como hemofilia B y afecta a uno de cada 30.000 varones. Según la Sociedad Española de Hematología y Hemoterapia (SEHH), en 2016 había en España alrededor de 3.000 pacientes con hemofilia (un 85% con hemofilia A y un 15% con hemofilia B); en el caso de la hemofilia A, el 96% de los casos se trata de varones y un 94% de los casos de hemofilia B.
Todas estas coagulopatías tienen una herencia autosómica recesiva. Clínicamente se expresan con hemorragias de intensidad variable, manifestándose una mayor gravedad en los casos homocigóticos, en los que existe una muy baja concentración de factor de coagulación. Los dos tipos principales de hemofilia (A y B) se transmiten de forma recesiva ligada al sexo (concretamente, en el cromosoma X del par 23), de tal forma que el 100% de las hijas de hemofílicos son portadoras y el 50% de los hijos de mujeres portadoras manifiestan la forma clínica de la hemofilia.
Existe un buen número de enfermedades genéticas ligadas a la pareja cromosómica sexual (XX en las mujeres y XY en los varones). Como los hombres la pareja es heterogénea (XY) sólo tienen una copia de los genes de cada uno de los dos cromosomas (X e Y), por lo que no existen otra copia del gen que puedan contrarrestar la función defectuosa del alelo anómalo situado en dicho cromosoma. Este es el caso de las hemofilias.
La inversión en la secuencia de ADN que se encuentra en el intrón 22 del gen del factor VIII es la anomalía molecular más frecuentemente detectada en pacientes con hemofilia A grave, siendo responsable de la enfermedad en el 45% de los casos. Aproximadamente el 50% de hemofílicos con afección grave y la práctica totalidad de hemofílicos con diátesis hemorrágica moderada o leve presentan mutaciones puntuales. En cambio, en la hemofilia B las deleciones parciales o completas del gen representan el principal defecto molecular. La existencia de mutaciones puntuales es responsable de las formas variantes de enfermedad (alteraciones funcionales). Se han identificado un importante número de alteraciones moleculares responsables de la hemofilia B; en este sentido, el gen humano F9, responsable de la producción de Factor IX, está situado en la región subtelomérica del brazo largo del cromosoma X; tiene una extensión de 34 kb y se divide en ocho exones y siete intrones. El Factor IX se sintetiza exclusivamente en el hígado por los hepatocitos y se procesa durante su secreción en el torrente sanguíneo. El polipéptido maduro del Factor IX contiene tres dominios: Gla - péptido de activación - dominio catalítico. El péptido de activación es liberado durante la conversión de Factor IX activado, que es modificado mediante hidroxilación y por un proceso de carboxilación dependiente de vitamina K (Cuéllar, 2013).
La hemofilia B es el resultado de múltiples mutaciones en el gen F9, y los pacientes con este trastorno de la coagulación se clasifican como positivos para material de reacción cruzada (CRM), caracterizados por presentar una concentración normal de antígeno de Factor IX pero una actividad reducida del mismo; asimismo, hay pacientes con CRM reducido, con niveles reducidos de antígeno de Factor IX y niveles de actividad y CRM negativos. Los pacientes CRM-positivos generalmente tienen mutaciones sin sentido en regiones codificantes o mutaciones de empalme o de transcripción que dan lugar a cantidades reducidas de Factor IX. Por su parte, los pacientes CRM-negativos suelen tener mutaciones de diversa tipología. En la hemofilia B, a diferencia de la hemofilia A, hay una intensa correlación entre la presencia de deleciones parciales o completas de genes y el desarrollo de inhibidores.
La expresión clínica más característica de todas las formas de hemofilia es la hemorragia, que se manifiesta en múltiples niveles y localizaciones orgánicas: muscular, sistema nervioso central, partes blandas y, muy especialmente, en las articulaciones. La gravedad de las manifestaciones clínicas suele estar en relación con la cantidad de factor existente:
- Forma grave. Cuando la actividad funcional del factor de la coagulación es indetectable, es decir inferior al 1%. Habitualmente se trata de pacientes con sangrado espontáneo antes de los 6 meses de edad o hemorragia intracraneal en el parto.
- Forma moderada. Los niveles de factor se encuentran entre el 1 y el 5% de lo normal. El sangrado aparece generalmente antes de los dos años de edad, tras producirse traumas mínimos o pequeñas maniobras exploratorias invasivas.
- Forma leve. Los niveles de factor VIII o IX son superiores al 5% e inferiores al 40%. El sangrado es raro y puede aparecer ante traumatismos importantes o tras intervenciones quirúrgicas.
La hemartrosis o hemorragia articular es la forma de sangrado más frecuente (65-90%), hasta el punto de que constituye el sello distintivo de las hemofilias. En pacientes con hemofilia A o B grave, más del 90% de todos los episodios hemorrágicos se producen en las articulaciones, y 80% de estos representan hemartrosis de los tobillos, rodillas y codos; es menos habitual en la articulación del hombro. En cualquier caso, la hemartrosis produce dolor, tumefacción e impotencia funcional.
Por detrás de la hemartrosis, en orden de frecuencia, los hematomas musculares suponen el 30% de las complicaciones hemorrágicas. Pueden complicarse con síndromes compartimentales e incluso shock hemorrágico; a la larga producen atrofia muscular. Finalmente, la hemorragia intracraneal es la complicación más grave, pero apenas constituye entre el 2 y 13% de las complicaciones hemorrágicas. Si no hay un tratamiento rápido puede causar la muerte. Los hemofílicos también pueden presentar complicaciones hemorrágicas en otras localizaciones, destacando la hematuria y la hemorragia gastrointestinal y orofaríngea.
La incidencia de sangrado en los pacientes con hemofilia durante el período neonatal oscila entre un 20% y un 44%, mientras que la hemorragia intracraneal (HIC) aparece en un 3,5-4% de los neonatos con hemofilia, aunque la cifra podría ser mayor si se incluyen las asintomáticas.
Hace ya más de 150 años que se conocen los beneficios de la transfusión de sangre para controlar la hemorragia relacionada con hemofilia y hace casi un siglo (1923), que Feissly demostrara la superioridad de plasma sobre la sangre total para este fin. La introducción en la década de 1970 de los concentrados con factores derivados del plasma permitió un tratamiento mucho más eficaz y funcional, así como una notable reducción de la morbilidad y mortalidad asociadas a la hemorragia.
Esta favorable evolución del tratamiento de restauración o de reemplazo de los factores sanguíneos implicados en las coagulopatías hereditarias sufrió a principios de la década de los años 80 del pasado siglo un grave revés. Nada menos que tres de cada cuatro pacientes con hemofilia grave acabaron infectados por el virus de la inmunodeficiencia humana (VIH) y prácticamente todos con el de la hepatitis C (VHC), como consecuencia de la transfusión de concentrados de plasma contaminados. En las décadas siguientes, la seguridad de estos productos se convirtió un elemento clave y derivó en la producción de concentrados no contaminados biológicamente, gracias a la incorporación de dobles sistemas de inactivación viral. A ello, cabe agregar la disponibilidad de análogos recombinantes de los factores naturales, carentes por completo de riesgo de contaminación por virus humanos.
Sin embargo, tanto los medicamentos de origen extractivo, a partir de plasma humano, como los de origen recombinante siguen teniendo un problema importante que puede limitar o incluso anular su utilidad: la producción de inhibidores en la sangre de los pacientes.
Los objetivos principales de la terapia son prevenir y, en su caso, tratar la hemorragia, así como sus complicaciones y secuelas, restaurar y mantener la función articular e integrar a los pacientes en la vida social normal. Para ello, los hemofílicos A moderados y leves se pueden tratar con desmopresina y antifibrinolíticos en un buen número de ocasiones; por el contrario, en las formas graves, es preciso utilizar un tratamiento sustitutivo con factor VIII (hemofilia A) o IX (hemofilia B), que pueden ser concentrados plasmáticos purificados o bien productos de origen recombinante. En todos los casos, la cantidad de factor a infundir depende de la gravedad de la hemorragia. Así, para combatir hemorragias graves o de riesgo vital se debe alcanzar un 100% de factor circulante. Ante una hemartosis, el tratamiento debe ser lo más precoz posible (antes de 4 horas) y ante la duda, siempre se debe tratar. El objetivo de factor a conseguir es de un 30-50% (habitualmente 20-40 U/kg de factor VIII y 30-60 U/kg de factor IX, según el tipo de hemofilia).
En recién nacidos con sospecha de hemofilia se recomienda administrar vitamina K por vía intravenosa a través de los vasos umbilicales. En caso de que no sea posible, otra alternativa es la vía oral, con una dosis de carga de 1 mg, seguida de 25 µg durante varios días.
La desmopresina es un análogo sintético de la vasopresina capaz de liberar del endotelio Factor vW vascular al torrente circulatorio, con aumentos de 4-5 veces su valor basal y durante 8-10 horas. Los efectos adversos son ligeros, habiéndose descrito enrojecimiento facial, cefalea, hipotensión y taquicardia. La desmopresina se administra por vía endovenosa o intranasal y debe considerarse como primera opción terapéutica en pacientes con hemofilia A leve y hemorragias leves o moderadas o procedimientos invasivos menores. Para las hemorragias mucocutáneas se ha mostrado especialmente útil la administración de ácido tranexámico.
La actividad de los factores de coagulación se expresa en unidades internacionales (UI), que corresponden al 100% de actividad del correspondiente factor en 1 ml de plasma de donantes sanos. La administración de 1 UI/Kg de peso aumenta 1-2% de su nivel en plasma. En general, los pacientes con enfermedad grave aumentan 1% después de la primera inyección mientras que el aumento del 2% solo se produce cuando se consigue un equilibrio entre los compartimentos intra y extravascular. En cualquier caso, los regímenes de dosificación habituales se basan en ajustes según el peso de los pacientes, pero la mejor comprensión de la respuesta farmacocinética de un individuo ha demostrado ser más eficaz en la predicción de los niveles de factor de coagulación que protegen contra episodios de sangrado.
El desarrollo de la artropatía hemofílica está directamente relacionado con el número de episodios de sangrado, aunque algunos estudios sugieren que el deterioro de las articulaciones puede ocurrir incluso sin evidencia clínica de hemartrosis. La artropatía, una vez establecida, es irreversible y progresiva; por lo tanto, la profilaxis, definida como la administración a largo plazo regular de los correspondientes factores implicados para prevenir hemorragias articulares, constituye el eje central de la gestión para los niños con hemofilia severa. La profilaxis primaria consiste en la infusión regular de concentrados del factor deficitario, que se mantiene durante más de 46 semanas al año, y es iniciada antes de la aparición de alteraciones articulares. Hay datos clínicos que demuestran que la profilaxis iniciada a temprana edad protege contra el daño de las articulaciones y disminuye la frecuencia de las hemartrosis y otras hemorragias.
ACCIÓN Y MECANISMO
El nonacog gamma es una forma recombinante del Factor IX de coagulación. Ha sido autorizada para el tratamiento y profilaxis de hemorragias en pacientes con hemofilia B (déficit de Factor IX). El Factor IX es un factor de coagulación dependiente de vitamina K que forma parte de la serina proteasas. Participa en la vía intrínseca de la coagulación sanguínea mediante la activación del factor X a su forma activa (Factor Xa), en presencia de calcio (Ca2+), fosfolípidos y cofactor VIII (complejo tenasa). El Factor X activado facilita la activación del Factor II (protrombina) a IIa (trombina) que, a su vez, activa la agregación plaquetaria y la conversión de Factor I (fibrinógeno) a Ia (fibrina); esta última es responsable de la estabilización del trombo sanguíneo mediante la formación de cadenas cruzadas de fibrina.
ASPECTOS MOLECULARES
El nanocog gamma es una forma recombinante del Factor IX de coagulación humano. Se trata de una glucoproteína de cada única formada por 415 aminoácidos. La molécula presenta varios dominios funcionales, incluyendo el dominio Gla, dos dominios de Factor de Crecimiento Epidérmico (EFG1 y EFG2), un péptido de activación y el dominio catalítico.
El proceso de activación del Factor IX consiste en la escisión de la cadena peptídica a nivel de los restos de arginina (Arg) de las posiciones 145 y 180, por la acción del Factor XIa (en la vía intrínseca de la coagulación) o de la combinación del Factor VIIa/Factor Tisular (en la vía extrínseca), lo que da lugar a dos cadenas peptídicas unidas por puentes disulfuro (-S-S-).
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del nonocog gamma han sido adecuadamente contrastadas en las indicaciones autorizadas mediante tres ensayos clínicos multicéntricos y abiertos.
El primero de ellos (250901; Winfiga, 2014) fue un estudio combinado de fases I/III, multicéntrico, prospectivo y controlado que incluyó a 73 pacientes con hemofilia B grave (<1% del nivel normal de Factor IX) o moderadamente grave (≤2%) y anteriormente tratados, con edades comprendidas entre los 12 y los 65 años. La equivalencia farmacocinética con el factor IX de origen extractivo fue confirmada en un subgrupo de 28 pacientes, expresada en términos de área bajo la curva concentración plasmática/tiempo (0-72 h) por dosis. La profilaxis mediante la administración de dos dosis por semana (media de 49,5 UI/kg por infusión IV) a lo largo de una media de seis meses fue efectiva para prevenir los episodios hemorrágicos, con una tasa anualizada de sangrados significativamente inferior (79%; p<0,001) a la obtenida en un control histórico utilizando una posología a demanda. Por otro lado, el 43% (24/56) de los pacientes tratados profilácticamente no experimentaron ninguna hemorragia durante todo el estudio. De los 249 episodios hemorrágicos experimentados durante el seguimiento, el 85% de ellos fueron controlados con una o dos infusiones IV de nonacog gamma. Globalmente, la eficacia hemostática en la resolución de los cuadros hemorrágicos fue calificada en el 96% de los casos como excelente (resolución completa del dolor y cese de los signos objetivos de hemorragia tras una única infusión IV) o buena (reducción sustancial del dolor y/o mejoría de los signos objetivos de hemorragia tras una única infusión IV, aunque requiriendo una segunda infusión para la resolución completa).
El segundo estudio (251101; EMA 2015) fue un ensayo clínico combinado de fases II/III, prospectivo no controlado y multicéntrico, realizado en 23 pacientes menores de 12 años (entre 1,8 y 11,8; mediana de 7,0) con hemofilia grave o moderadamente grave, que había recibido un tratamiento profiláctico con nonocog gamma durante seis meses con dos dosis semanales. Como variable primaria se determinó la incidencia de eventos adversos posible o problemente relacionados con el tratamiento, que fue del 0%. La mediana de la tasa anualizada de sangrados fue de 2,0 (2,0 para los menores de 6 años y 1,8 para 6-12 años), mientras que la eficacia hemostática global fue considerada excelente o buena en el 100% en los menores de 6 años y en el 93% entre 6 y 12 años; el número de infusiones de nonacog gamma necesarios para controlar cada episodio hemorrágico espontáneo fue de 1 o 2 en el 91% de los casos en menores de 6 años y en el 87% en el esto. La mediana del consumo anualizado de nonacog gamma fue de 4,72 UI/kg entre los menores de 6 años y de 4,53 UI/kg en el resto.
El último estudio (251002; EMA 2015) es un ensayo de fase III, prospectivo, abierto, no controlado y multicéntrico, cuyo objetivo es evaluar la eficacia y la seguridad del nonocog en 14 sujetos mayores de 16 años con hemofilia B grave o moderadamente grave que fueron sometidos a procedimientos de cirugía mayor (n=11) o intervenciones menores, como cirugía dental o artroscopia (n=3). La variable clínica primaria fue consistió en la valoración de la eficacia hemostática intraoperatoria, que resultó excelente en el 100% de los casos, tanto de cirugía mayor como menor. La mediana de pérdida de sangre intraoperatoria fue de 100 ml entre los sometidos a cirugía mayor y de 2 ml en cirugía menor. La eficacia hemostática posoperatoria fue valorada tras el alta hospitalaria como buena o excelente en todos los casos (salvo un paciente de cirugía menor, que no fue valorado en esta variable).
Desde el punto de vista de la seguridad, el nonocago gamma presenta un perfil toxicológico benigno, equiparable al de otros factores de coagulación utilizados en clínica. De todos los pacientes incluidos en el programa de investigación clínica (n=99) solo se manifestaron disgeusia en un paciente (0,014% considerando todas administraciones IV realizadas) y dolor en las extremidades en otro (0,007%).
Por el momento, no se ha observado el desarrollo de inhibidor tras la administración de nonocag gamma, aunque teóricamente es posible tal formación; en el caso de que así sucediese, previsiblemente se manifestaría como una merma de la respuesta hemostática. En este sentido, parece existir una cierta relación entre el desarrollo de inhibidor de Factor IX y la aparición de reacciones alérgicas, en incluso reacciones anafilácticas, por lo que la posible aparición de cuadros alérgicos podría sugerir el desarrollo de inhibidor.
ASPECTOS INNOVADORES
El nonacog gamma es una forma recombinante del Factor IX de coagulación. Ha sido autorizada para el tratamiento y profilaxis de hemorragias en pacientes con hemofilia B (déficit de Factor IX). Los datos acumulados de los tres estudios clínicos disponibles han demostrado la equivalencia farmacocinética del nonacog gamma con respecto al factor IX de origen extractivo; asimismo, se han establecido adecuadamente su eficacia y su seguridad, tanto en uso profiláctico como a demanda, incluyendo pacientes pediátricos y sometidos a intervenciones quirúrgicas. Aparentemente, no hay diferencias entre el nonacog gamma y los medicamentos con factor IX de origen extractivo disponibles actualmente, ya que a estas alturas las exigencias de seguridad y calidad de estos últimos son lo suficientemente elevadas como para que tal origen condicione su uso clínico.
|
VALORACIÓN |
|
NONACOG GAMMA
|
|
Grupo Terapéutico (ATC): B02BD. SANGRE Y ÓRGANOS HEMATOPOYÉTICOS. Vitamina K y otros hemostáticos: factores de la coagulación sanguínea. |
|
Indicaciones autorizadas: Tratamiento y profilaxis de hemorragias en pacientes con hemofilia B (deficiencia congénita de factor IX). |
|
sin INNOVACIÓN. No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas. |

|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Factor IX |
Mononine |
UCB CSL Behring |
1994 |
|
Factor IX Grifols |
UC Grifols B |
2004 |
|
|
Immunine |
Baxalta |
2009 |
|
|
Octanine |
Octapharma |
2011 |
|
|
Nonacog gamma |
Rixubis |
Baxalta |
2016 |
BIBLIOGRAFÍA
Bibliografía
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- European Medicines Agency (EMA). Rixubis®. European Public Assessment Report (EPAR). EMA/663763/2014; EMEA/H/C/003771. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003771/WC500182068.pdf
- Cuéllar Rodríguez S. Hemofilia y otras coagulopatías hereditarias. Panorama Actual Med. 2013; 37(362): 232-42.
- Cuéllar Rodríguez S. Efmoroctocg alfa (Eloctin) en hemofilia A. Panorama Actual Med. 2016; 40(398): 1012-7.
- Turecek PL, Abbühl B, Tangada SD, Chapman M, Gritsch H, Rottensteiner H, et al. Nonacog gamma, a novel recombinant factor IX with low factor IXa content for treatment and prophylaxis of bleeding episodes. Expert Rev Clin Pharmacol. 2015; 8(2): 163-77. doi: 10.1586/17512433.2015.1011126.
- Windyga J, Lissitchkov T, Stasyshyn O, Mamonov V, Rusen L, Lamas JL, et al. Pharmacokinetics, efficacy and safety of BAX326, a novel recombinant factor IX: a prospective, controlled, multicentre phase I/III study in previously treated patients with severe (FIX level <1%) or moderately severe (FIX level ≤2%) haemophilia B. Haemophilia. 2014; 20(1): 15-24. doi: 10.1111/hae.12228.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
