Ivacaftor Kalydeco® (Vertex)
 Nº386
Nº386

Resumen
El ivacaftor es un potenciador de la proteína CFTR, es decir, incrementa la actividad del canal de cloruro (CFTR) debido a un aumento del tiempo durante el cual el canal permanece abierto. Ha sido autorizado como medicamento huérfano para el tratamiento de pacientes con fibrosis quística de 6 años de edad y mayores con una de las siguientes mutaciones de apertura del canal (clase III) en el gen CFTR: Gly551Asp (G551D), Gly1244Glu (G1244E), Gly1349Asp (G1349D), Gly178Arg (G178R), Gly551Ser (G551S), Ser1251Asn (S1251N), Ser1255Pro (S1255P), Ser549Asn (S549N) o Ser549Arg (S549R). La proteína CFTR es un canal de cloruro (Cl–) regulado por AMPc, que se localiza en la membrana de las células epiteliales, donde actúa regulando el flujo de electrolitos y de agua. Su disfunción – de origen genético – modifica la cantidad y la composición relativa de los fluidos, afectando a los órganos con tejido epitelial, provocando una excesiva absorción de fluidos y la correspondiente deshidratación de la superficie epitelial, que es responsable de la formación de un moco hiperviscoso (mucoviscidosis) con un aumento de la concentración mucina polimérica, característica de la fibrosis quística y responsable de la mayor parte de los trastornos clínicos ligados a la enfermedad.
FIBROSIS QUÍSTICA (MUCOVISCIDOSIS)
La fibrosis quística o mucoviscidosis es una enfermedad crónica, degenerativa y hereditaria que afecta principalmente a las zonas del cuerpo que producen secreciones mucosas y, en particular, a los pulmones y al aparato digestivo, dando lugar a un incremento de la viscosidad de dichas secreciones como consecuencia de la disminución de su contenido de agua, sodio y potasio. La consecuencia primaria de ello es la obstrucción de los canales que transportan dichas secreciones, permitiendo que el correspondiente estancamiento facilite el desarrollo de infecciones e inflamaciones que destruyen las zonas implicadas, especialmente pulmón, hígado, páncreas y sistema reproductor. Es una patología con una esperanza de vida limitada y que por el momento no tiene curación (FEFQ, 2014).
La fibrosis quística es el trastorno genético más común entre individuos de raza blanca, aunque su incidencia es muy variable incluso dentro de cada país. En Europa se estima globalmente en 1:8.000 individuos, yendo desde 1:1.500 en Irlanda hasta 1:25.000 en Finlandia, con incidencias entre 1:2.000 y 1:4.000 para Suecia, Francia, Italia, Holanda, Suiza y Gran Bretaña. En España, es de un caso de cada 5000 nacidos vivos (1:5.000), aunque uno de cada 35 habitantes son portadores sanos de la enfermedad. Esta incidencia global convierte a la fibrosis quística en una enfermedad rara, aunque se trata sin duda de la más común de esta categoría1.
Es una enfermedad crónica y progresiva, que suele aparecer durante la infancia temprana o, más raramente, en el nacimiento (íleo meconial). Aunque un cierto número de casos son diagnosticados durante la fase adulta de la vida, cada vez es menos frecuente debido al establecimiento de pruebas sistemáticas de diagnóstico perinatal.
En principio, cualquier órgano interno puede verse afectado, aunque las principales manifestaciones afectan al aparato respiratorio (bronquitis crónica y exacerbaciones), al páncreas (insuficiencia pancreática, diabetes del adolescente y ocasionalmente pancreatitis) y, más raramente, al intestino (obstrucción) o al hígado (cirrosis). La forma más común de fibrosis quística se asocia a síntomas respiratorios, a problemas digestivos (esteatorrea y/o estreñimiento) y a anomalías del crecimiento. Es característico de esta enfermedad el sabor salado de la piel.
La importante morbimortalidad de esta enfermedad está relacionada con la afectación pulmonar y sus complicaciones que son responsables del 95% de los fallecimientos de los pacientes que la padecen (MSC, 2010). Aunque los pulmones del recién nacido con fibrosis quística son histológicamente normales, en los primeros meses de la vida algunos pacientes comienzan a desarrollar una colonización crónica de la tráquea por determinadas bacterias, como consecuencia de la hiperviscosidad del moco que provoca obstrucción bronquial y una respuesta inflamatoria. Todo ello constituye un círculo vicioso que evoluciona hacia una lesión irreversible del pulmón.
Las bacterias colonizadoras patógenas más comúnmente encontradas en los pacientes con fibrosis quística son Pseudomonas aeruginosa, Sthapylococcus aureus, Hemophilus influenzae, Stenotrophomonas maltophilia, Achromobacter xylosoxidans y Burkholderia spp. De todos ellas, es sin duda Pseudomonas aeruginosa la más relevante, tanto por su frecuencia como por su potencial patogénico y su resistencia natural frente a muchos agentes antibacterianos. Esta bacteria penetra en el tracto respiratorio inferior por inhalación y puede infectar transitoriamente las vías aéreas en algunos pacientes con fibrosis quística (10-50%). Aunque algunos de los pacientes infectados parecen ser capaces de eliminar esta colonización sin ninguna ayuda, en la mayoría de los casos persiste en forma de infección crónica, con eventuales reagudizaciones o exacerbaciones (Döring, 2012).
La infección crónica da lugar a una respuesta inflamatoria prolongada, a la que se atribuye el daño pulmonar característico de la enfermedad que acaba por manifestarse como una deficiente función respiratoria y, de hecho, los datos clínicos demuestran que la erradicación precoz de la infección pulmonar y la prevención de la infección crónica se asocia a un mejor pronóstico de la fibrosis quística.
La mayoría de los pacientes tienen un deterioro respiratorio insidioso con exacerbaciones de su infección pulmonar crónica manifestadas por aumento de la tos, cambios en el volumen y en el aspecto del esputo, aparición de disnea, disminución de la actividad física, pérdida de peso y de apetito, cambios en la auscultación habitual, imágenes radiológicas nuevas y, sobre todo, deterioro de la función pulmonar. En este sentido, la disminución del volumen pulmonar o atelectasia se presenta en el 5-10% de los pacientes, especialmente adultos, que se debe a la formación de tapones de secreciones espesas o como complicación de una aspergilosis broncopulmonar alérgica (ABPA), una reacción de hipersensibilidad al Aspergillus fumigatus que se presenta entre un 10-20% de los pacientes.
El 60% de los pacientes adultos presentan una leve hemoptisis de manera recurrente, generalmente asociada a exacerbaciones pulmonares. Un 10% presenta neumotórax, aunque esta proporción aumenta con la edad, generalmente por la ruptura de bullas subpleurales de la pleura visceral, presentándose como un cuadro de dolor torácico brusco y dificultad respiratoria. La insuficiencia respiratoria es un signo de enfermedad pulmonar avanzada. La reducción de la presión parcial de oxígeno en la sangre arterial por debajo de 80 mmHg (hipoxemia) suele preceder al aumento de la de dióxido de carbono por encima de 46 mmHg (hipercapnia), la cual produce acidosis y estimula los movimientos respiratorios.
Las manifestaciones clínicas gastrointestinales más relevantes están relacionadas con el grado de afectación del páncreas, siendo los síntomas clínicos más destacados la diarrea (particularmente, esteatorrea), el retraso del desarrollo y la desnutrición. Entre un 7% y un 10% de los recién nacidos2 con fibrosis quística presentan íleo meconial. En condiciones normales, el meconio está formado por un amasijo espeso de células muertas y secreciones del estómago y del hígado, el cual reviste el intestino del recién nacido y, de hecho, formas las primeras heces que éste expulsa. En el caso del íleo meconial, este meconio es especialmente viscoso, debido a la insuficiencia de enzimas pancreáticos, provocando la oclusión y, eventualmente, el estallido intestinal. El íleo meconial puede ser causa de estenosis e incluso atresia intestinal, aunque lo más frecuente es que se produzca en las primeras 24-48 horas de vida extrauterina ocasionando la no emisión de meconio, distensión abdominal y vómitos.
El síndrome de obstrucción intestinal distal (SOID, DIOS en inglés) presenta una fisiopatología análoga a la del íleo meconial, pero se produce en edades más avanzadas, incluso en adolescentes y adultos, afectando al 20 % de los pacientes con fibrosis quística. Provoca, dentro de un cuadro de estreñimiento agudo, una oclusión del íleon terminal y/o colon ascendente, lo que se manifiesta como una gran distensión abdominal. El vólvulo intestinal3 se presenta con más frecuencia durante la lactancia y se debe en buena medida a la viscosidad de las heces. En el caso de ocurrir durante la vida fetal puede provocar atresia o estenosis o íleo meconial complicado.
Muchos pacientes (en torno al 30%) presentan enfermedad por reflujo gastroesofágico (ERGE), eventualmente asociada a microaspiraciones bronquiales del contenido gástrico que complican aún más la patología respiratoria, aumentando su incidencia con la edad. Mucho menos común es la colonopatía fibrosante, que posiblemente está relacionada con el consumo de grandes cantidades de enzimas para combatir la insuficiencia pancreática, particularmente en varones, así como en pacientes con íleo mecánico y en aquellos que toman laxantes o antiinflamatorios no esteroideos frecuentemente.
La secreción exocrina del páncreas – conteniendo bicarbonato y diversos enzimas, como amilasa, proteasa y lipasa – está muy limitada en la fibrosis quística, hasta el punto de que la insuficiencia pancreática llega a afectar a más del 85% de los pacientes. Esta insuficiencia pancreática produce, entre otras cosas, procesos de malabsorción y ésta se relaciona – en particular con las grasas no absorbidas – con la esteatorrea (diarrea grasa). Esta malabsorción de grasas disminuye correlativamente la de las vitaminas liposolubles (A, D, E y K, principalmente). Además, en los pacientes con fibrosis quística, debido a la esteatorrea, las pérdidas fecales de ácidos biliares aumentan notablemente. En pacientes adolescentes o adultos también se presenta síntomas y signos de pancreatitis, aunque no es habitual (menos del 1%). En cualquier caso, los pacientes con fibrosis quística presentan un deterioro progresivo del páncreas, manifestándose con una reducción de la tolerancia a la glucosa, que desemboca con cierta frecuencia (por encima del 8% a partir de los 10 años de edad) en un cuadro de diabetes mellitus.
La lesión hepática se relaciona con la obstrucción de los conductos biliares intrahepáticos por una bilis espesa. Debido a la comunicación que existe entre los conductos biliares intrahepáticos, el bloqueo de algunos de ellos no produce necesariamente un cuadro de colestasis completa ni da lugar en la mayoría de los pacientes a signos clínicos o bioquímicos de alteración hepática. Sin embargo, hay una evolución fibrótica silente que llega a producir la atrofia del parénquima hepático, con desarrollo final de una cirrosis multilobular. Finalmente, la prevalencia de litiasis biliar es algo más frecuente en pacientes con fibrosis quística que en individuos sanos, alcanzando porcentajes de un 12 % en niños y adultos, y aunque suele ser asintomática, puede llegar a requerir cirugía.
Los pacientes varones con fibrosis quística suelen presentar anomalías anatómicas en su aparato reproductor. Los testículos son de menor tamaño o se encuentran en una situación anormal, generalmente ocultos en el abdomen (criptorquidia). Los conductos deferentes o bien no existen (ABCD: Ausencia Bilateral de Conductos Deferentes) o están atrofiados; asimismo, suele haber ausencia o atrofia del cuerpo y de la cola del epidídimo, y en un importante número de casos hay atrofia de las vesículas seminales. También existe azoospermia (ausencia de espermatozoides) en el 95-98% de los pacientes varones, por lo que estos son estériles; además, su volumen de eyaculado puede estar reducido y el pH puede ser más ácido de lo normal. Pese a ello, los niveles hormonales en los pacientes son normales.
A diferencia del varón, la mujer con fibrosis quística no presenta anomalías en su aparato reproductor, aunque suelen presentar una pubertad y una menarquia retrasadas en torno a dos años, si se las compara con la población sana. Presentan con frecuencia alteraciones menstruales, con periodos de amenorreas, aunque posiblemente secundarias a la desnutrición que pueden presentar algunas pacientes. La fertilidad femenina, a diferencia de la masculina, se encuentra conservada, aunque mermada, estimándose en un 10-20% de la mujer sana.
El característico sabor salado de la piel del lactante con fibrosis quística es debido a aumento en la concentración de sal (cloruro sódico, ClNa) en el sudor. En condiciones normales, el sudor es secretado en el acino de la glándula sudorípara como una solución isotónica con el plasma; el ducto de la glándula sudorípara es impermeable al agua y, en condiciones normales, existe una reabsorción activa de cloruro y de sodio que hace que el sudor sea hipotónico en sujetos normales, con bajas concentraciones salinas. En los pacientes con fibrosis quística, la proteína implicada (CFTR) es el denominado regulador de la conductancia transmembrana, un canal de cloruro que se expresa en las células epiteliales de los órganos en los que se manifiesta la enfermedad, entre ellos los ductos de las glándulas sudoríparas. La activación del canal produce normalmente su apertura, y en el caso del ducto de la glándula sudorípara, la reabsorción del cloruro y sodio para producir el sudor hipotónico normal. Por el contrario, en los pacientes con fibrosis quística el CFTR no existe, no está correctamente situado en la membrana celular o es anómalo y no responde adecuadamente a su estímulo fisiológico, o bien sus propiedades conductoras del cloruro y de sodio son inferiores a lo normal.
Las altas concentraciones salinas en el sudor de los pacientes pueden producirles síndromes clínicos, típicamente asociados a los periodos de tiempo caluroso, la postración con el calor, una forma de deshidratación hiponatrémica aguda con hipotensión. Sin embargo, es más frecuente la alcalosis hipoclorurémica crónica, que se origina por la depleción salina de una manera menos abrupta y con síntomas subagudos de apatía o irritabilidad, rechazo de la alimentación, vómitos y falta de ganancia de peso.
El diagnóstico se sospecha por los resultados de la prueba del sudor (concentración del cloruro por encima de 60 mmol/L) y se confirma con la identificación de alguna mutación del gen CFTR. El diagnóstico neonatal mediante cribado está disponible desde el año 2002 y ha permitido diagnosticar el 95% de los nuevos casos registrados. Su importancia es capital, ya que un diagnóstico precoz de la fibrosis quística permite prolongar la esperanza de vida de los pacientes y mejorar sustancialmente su calidad; por otro lado, la técnica es extremadamente sencilla y con escasos costes económicos. Hay también pruebas prenatales, que se realizan mediante el análisis de mutaciones sobre muestras de vellosidades coriónicas obtenidas durante la octava semana de gestación. Asimismo, las técnicas de fecundación in vitro combinadas con la tecnología de la reacción en cadena de la polimerasa permite la detección de anomalías genéticas mediante el diagnóstico genético preimplantacional. Éste puede realizarse en ovocitos o en blastómeros de embriones en estadios previos a la implantación. Este tipo de análisis es especialmente recomendado a parejas portadoras de una mutación que hayan tenido un hijo con fibrosis quística anteriormente. Además, es aconsejable el consejo genético para cualquier pareja portadora de mutaciones en heterocigosis (identificadas tras el nacimiento de un primer niño con fibrosis quística, por antecedentes familiares de la enfermedad o tras la detección al nacer de una mutación heterocigótica del recién nacido).
El test del sudor sigue siendo esencial para efectuar el diagnóstico de la fibrosis quística en jóvenes y adultos, tanto por su fiabilidad como por su bajo coste. Se considera confirmado el diagnóstico tras dos pruebas positivas; no obstante, la concentración – siempre altos – de cloruro en sudor no está directamente relacionada con el nivel de gravedad clínica de la enfermedad. La confirmación definitiva viene tras la determinación, en su caso, de una o más mutaciones en el gen CFTR. En general, en España los pacientes diagnosticados de fibrosis quística durante la edad adulta suelen presentar una disfunción respiratoria y pancreática menor que las formas diagnosticadas neonatalmente o durante la infancia, y el pronóstico tiende a ser más favorable (Lerín, 2014).
La mortalidad y morbilidad asociada a la fibrosis quística dependen fundamentalmente del grado de afectación broncopulmonar. La esperanza de vida se ha incrementado notablemente, pasando de ser menos de cinco años hace medio siglo a alcanzar los 40 años actualmente.
La fibrosis quística se caracteriza por alteraciones en la proteína CFTR o regulador de la conductancia transmembrana de fibrosis quística (cystic fibrosis transmembrane conductance regulator,) que juega un papel determinante en la regulación del flujo hidroelectrolítico a través de la membrana en numerosos tipos de células (Bellón, 2006). Estas alteraciones provocan cambios en las características de las secreciones de las diversas glándulas exocrinas existentes en el organismo humano, haciéndolas notablemente más viscosas; de ahí el término mucoviscidosis con el que también es conocida la fibrosis quística.
Se trata de una enfermedad congénita genéticamente determinada por la existencia de mutaciones en el gen que codifica la proteína CFTR, localizado en el cromosoma 7. Hasta el momento se han descrito más de 2.000 mutaciones diferentes de dicho gen que pueden estar relacionadas con la fibrosis quística (Ehre, 2014), aunque la mayor parte de los casos se asocian con la mutación F508del; no obstante, no hay una correlación clara entre el genotipo y el fenotipo. Por otro lado, además de la heterogeneidad alélica y de la posible ocurrencia de mutaciones múltiples en el mismo gen, existe una amplia gama de factores puede influir en la expresión clínica (fenotipo) de la enfermedad, incluyendo el medio ambiente, el estilo de vida y la presencia otros diversos genes, mutados o no, que pueden condicionar el fenotipo.
La herencia genética de la fibrosis quística es de carácter autosómico recesivo, es decir, solo se llega a manifestar la enfermedad si los dos alelos del gen CFTR recibidos del padre y de la madre están alterados, mientras que si se hereda un alelo normal y uno defectuoso se adquiera la condición de portador sano de la enfermedad, con la posibilidad de transmitirla a la descendencia. El riesgo teórico para la descendencia para cada concepción en una pareja en la que ambos sean portadores es del 25% de hijo sano, del 50% de portador y del 25% de estar afectado por la enfermedad.
La proteína CFTR es un canal de cloruro (Cl–) regulado por AMPc, que requiere ATP y la fosforilación del dominio regulador para su funcionamiento. Forma parte de la denominada superfamilia ABC de transporte de proteínas (ATP-Binding Cassette transporter superfamily). Además de controlar la secreción de cloruro, este canal regula la función de otras proteínas de membrana, incluyendo el canal epitelial de sodio (ENaC). También juega un papel importante en la secreción celular de bicarbonato (HCO3–), un electrolito alcalinizante que juega un papel crucial en el mantenimiento del pH y, en consecuencia, en la capacidad de respuesta frente a los microorganismos, en particular frente a las bacterias.
La proteína CFTR se localiza en la membrana de las células epiteliales, donde actúa regulando el flujo de electrolitos y de agua. Si dicho canal se encuentra alterado o no existe, como consecuencia de alguna mutación del gen CFTR, se modifica la cantidad y la composición relativa de los fluidos, afectando a los órganos con tejido epitelial. En definitiva, la disfunción del CFTR provoca una excesiva absorción de fluidos y la correspondiente deshidratación de la superficie epitelial, que es responsable de la formación de un moco hiperviscoso con un aumento de la concentración mucina polimérica. (Ehre, 2014).
Entre otros tejidos, el CFTR es expresado fundamentalmente en páncreas, intestino, glándulas sudoríparas, pulmones y tracto genital. En las glándulas sudoríparas de personas sanas, el flujo celular de Cl– va en paralelo con el gradiente de Na+, reabsorbiéndose ambos electrolitos a través de la membrana apical de las células del conducto glandular. En ausencia o deficiente funcionamiento del CFTR se produce un aumento de la concentración de ClNa en el sudor, dando lugar al característico sabor salado en los pacientes con fibrosis quística, hasta el punto de constituir un elemento clave para el diagnóstico.
También la alteración o ausencia del CFTR repercute sobre distintos mecanismos de defensa en las vías respiratorias. La actividad antimicrobiana de algunas proteínas (lisozima, lactoferrina) se ve reducida cuando la concentración de ClNa es alta, ya que impide su unión a las bacterias favoreciendo las infecciones. También se postula que la motilidad ciliar disminuye a alta concentración de Na+ impidiendo la eliminación del moco. Por su parte, en la membrana apical de los conductos pancreáticos de las personas sanas, los canales de Cl– y Cl–/HCO3– funcionan en paralelo. En condiciones normales, el HCO3– (bicarbonato) es liberado en el lumen produciendo una secreción alcalina que hidrata, solubiliza e inactiva las secreciones acinares, pero en los pacientes con fibrosis quística este proceso se altera y las secreciones espesas obstruyen los conductos y esto provoca la destrucción del páncreas. De forma similar, la alteración de los canales de Cl–, Na+ y K+ en el intestino son la causa de la deshidratación y compactación que pueden ocasionar íleo meconial y síndrome de obstrucción intestinal distal (DIOS). También se ha sugerido que la alteración del canal CFTR sería la causa de la esterilidad masculina debido al taponamiento de los conductos deferentes.
El gen CFTR fue identificado en 1989 en la región 7q31 del cromosoma 7. Está formado por unas 250.000 bases nucleicas (250 kb) y contiene 27 zonas codificantes (exones) que codifican una proteína de 1.480 aminoácidos, la proteína CFTR. Ésta está formada por dos dominios transmembrana (membrane-spanning domains, MSD), cada uno de los cuales está unido a un dominio citoplasmático de unión a nucleótidos (nucleotid binding domain, NBD), estando unidos estos dos módulos de MSD-NBD por un único dominio citoplasmático regulador (regulatory, R), con múltiples puntos de fosforilación para las cinasas A y C, lo que facilita que el canal sea regulado fisiológicamente por hormonas que actúan mediante dichas cinasas. En este sentido, la fosforilación del dominio R es una prerrequisito para la actividad del canal; en concreto, tras la fosforilación, el proceso de control de su apertura y cierre viene determinado por la unión de ATP y la hidrólisis en un dímero de NBD.
La disfunción de la proteína CFTR se produce por distintos mecanismos moleculares en base a los cuales las mutaciones se agrupan en seis categorías, aunque algunas mutaciones pueden incluirse en más de una clase.
Las mutaciones de clase I afectan la biosíntesis de CFTR. Impiden la síntesis de una proteína estable debido a una mutación sin sentido o producen de una proteína truncada debido a la creación de un codón de parada prematuro. Estas proteínas truncadas son inestables y se degradan rápidamente por las proteínas en el retículo endoplásmico. Las mutaciones de Clase I incluyen los fenotipos más graves, porque ninguna proteína funcional se sintetiza. Este tipo de mutación se encuentra en el 5% -10% de los cromosomas CF en todo el mundo, pero representan alrededor del 60% de las mutaciones en pueblo judío con fibrosis quística CF. La mutación sin sentido se indica terminando con una X y los ejemplos más conocidos son Trp1282X (anteriormente W1282X)4, Arg1162X (R1162X) y Gly542X (G542X).
Las de clase II afectan la maduración de la CFTR, dando lugar a un inadecuado procesamiento de la proteína y hacen que la CFTR esté mal plegada, impidiendo su ubicación correcta en la membrana celular apical, lo que facilita su degradación prematura por el proteasoma; además, las pocas moléculas que alcanzan esta ubicación, funcionan incorrectamente. La mutación supresora Phe508del explica la mayoría de las mutaciones en este grupo. De hecho, alrededor del 90% de las personas con fibrosis quística son heterocigotos y un 40-50% son homocigotos para el Phe508del.
Las mutaciones de clase III afectan la regulación del canal o compuerta de cloruro. La producción de la proteína CFTR y la migración hacia la membrana son normales, pero cuando la proteína llega a la superficie de la membrana no responde a la estimulación de AMPc. Esto impide la unión de ATP y/o la fosforilación. La proteína CFTR no se abre en respuesta a señales intracelulares en la superficie celular y permanece cerrado la mayor parte del tiempo. La mutación de sentido erróneo Gly551Asp (G551D) es la tercera mutación CFTR más común y causa una sustitución de glicina (Gly) por aspartato (Asp) en la posición 551 de la proteína. Un 4-5% de todos los pacientes con fibrosis quística tienen la mutación Gly551Asp en al menos un alelo, aunque esta mutación es más frecuente en las personas de origen celta. La mutación Gly551Asp (G551D) se considera grave, no así la Ala455Glu (A455E), también de clase III.
La clase IV agrupa a las mutaciones que determinan un flujo reducido de iones cloruro a través del canal. Se debe a que éste adopta una conformación anómala, impidiendo una permeación adecuada de los iones cloruro y reduciendo la probabilidad de apertura del canal; por lo general se trata de mutaciones leves Arg117His (R117H) es la mutación más común de clase IV, con una frecuencia de 2-4%.
Las mutaciones de clase V afectan la estabilidad del ARNm que permite transcribir la proteína CFTR. La maquinaria de empalme se altera, lo que resulta en la generación de cadenas de ARNm tanto aberrantes como normales, lo que conduce a un número reducido de proteínas CFTR funcionales en la membrana plasmática. El ejemplo característico es la mutación 3849+10kbC<T, presente en menos de 1% de los pacientes con fibrosis quística. El nivel de CFTR funcionamiento puede variar entre diferentes pacientes e incluso entre los diferentes órganos del mismo paciente y a menudo resulta en un fenotipo clínico más leve.
Finalmente, las mutaciones de clase afectan la estabilidad de la CFTR. El truncamiento (X) del extremo C (carboxi-terminal) de CFTR produce una proteína funcional pero que es inestable en la superficie de la membrana apical; su ejemplo mejor conocido es la Gln1412X (Q1412X).
Como se ha indicado, la mutación más frecuente es de clase II se debe a la pérdida (deleción) del aminoácido fenilalanina (Phe, F) en el codón 508 (Phe508del, F508del) de la proteína CFTR. Esta mutación afecta globalmente en España a los cromosomas del 52% de los pacientes con fibrosis quística; sin embargo, se ha observado una frecuencia muy diversa a lo largo de la geografía española: País Vasco 87%, Asturias 78%, Castilla y León 65%, Aragón 58%, Cataluña 50%, Galicia 45%, Andalucía 43% y Murcia 34% (Alonso, 2007). Por su parte, la mutación G551D es la más frecuente de las mutaciones de clase III e implica la sustitución de glicina (G, Gly) por ácido aspártico (D, Asp) en la posición 551 de la proteína CFTR, lo que se traduce en una reducción en la actividad del canal, debida a un acortamiento del tiempo durante el cual el canal permanece abierto. De las más de 2.000 mutaciones identificadas para el gen CFTR, en España se han identificado alrededor de 100, de las cuales poco más de diez presentan una frecuencia superior al 1% y acumulan las tres cuartas partes de los casos. Además de la F508del (51,7% de los casos registrados en España), las más comunes son Gly542X (G542X; 7,7%), Asn1303Gln (N1303Q; 2,9%), 1811+16kbA>G (1,8%) y Arg344Trp (R344W; 1,8%).
En general, el fenotipo de insuficiencia pancreática está asociado a la presencia de dos mutaciones graves, mientras que la correlación con la enfermedad pulmonar es mucho más compleja, posiblemente por la mayor incidencia de otros factores, tanto ambientales como genéticos. En general, las mutaciones leves (R117H, L206W,) determinan la aparición tardía de los síntomas, cuando el niño tiene ya varios años o incluso en la adolescencia o en la fase adulta de la vida.
A pesar del desarrollo incipiente de fármacos susceptibles de actuar sobre algunos de los efectos celulares de la enfermedad y en particular sobre la alteración del regulador de conductancia transmembrana (CFTR), aún estamos lejos de disponer de un tratamiento auténticamente eficaz para afrontar las causas genéticas y moleculares de la fibrosis quística, controlar sus manifestaciones clínicas más relevantes y prevenir la progresión del deterioro orgánico provocado por la enfermedad.
El tratamiento actual es fundamentalmente sintomático y de mantenimiento de las funciones fisiológicas, particularmente a nivel respiratorio, metabólico y digestivo, así como la prevención y tratamiento de los cuadros infecciosos (en particular, los pulmonares). A pesar de sus limitaciones y complicaciones, estos tratamientos han mejorado notablemente la esperanza de vida de estos pacientes, pasando de menos de cinco años hace medio siglo a los 40 que se estiman actualmente; esto implica, de hecho, que un número creciente de pacientes llegará alcanzar edades avanzadas – equiparables al promedio del resto de personas – en un futuro esperanzadoramente cercano, particularmente con la investigación en curso de tratamientos etiológicos eficientes (incluyendo las nuevas terapias avanzadas: terapia génica, terapia celular e ingeniería tisular), pruebas sistemáticas pre y neonatales, y la consolidación definitiva de un amplio número de centros de referencia (Cuéllar, 2014).
ACCIÓN Y MECANISMO
El ivacaftor es un potenciador de la proteína CFTR, es decir, incrementa la actividad del canal de cloruro (CFTR) debido a un aumento del tiempo durante el cual el canal permanece abierto. Ha sido autorizado para el tratamiento de pacientes con fibrosis quística de 6 años de edad y mayores con una de las siguientes mutaciones de apertura del canal (clase III) en el gen CFTR: Gly551Asp (G551D), Gly1244Glu (G1244E), Gly1349Asp (G1349D), Gly178Arg (G178R), Gly551Ser (G551S), Ser1251Asn (S1251N), Ser1255Pro (S1255P), Ser549Asn (S549N) o Ser549Arg (S549R).
La proteína CFTR es un canal de cloruro (Cl–) regulado por AMPc, que requiere para su funcionamiento ATP y la fosforilación del dominio regulador. Se localiza en la membrana de las células epiteliales, donde actúa regulando el flujo de electrolitos y de agua. Cuando este canal se encuentra alterado o no existe, como consecuencia de alguna mutación del gen CFTR, se modifica la cantidad y la composición relativa de los fluidos, afectando a los órganos con tejido epitelial. En definitiva, la disfunción del CFTR provoca una excesiva absorción de fluidos y la correspondiente deshidratación de la superficie epitelial, que es responsable de la formación de un moco hiperviscoso (mucoviscidosis) con un aumento de la concentración mucina polimérica, característica de la fibrosis quística.
Por ahora no se conoce con detalle cuál es el mecanismo íntimo de acción por el que el ivacaftor incrementa dicho tiempo de apertura de la proteína CFTR. Ésta está formada por dos dominios transmembrana (membrane-spanning domains, MSD), cada uno de los cuales está unido a un dominio citoplasmático de unión a nucleótidos (nucleotid binding domain, NBD), estando unidos estos dos módulos de MSD-NBD por un único dominio citoplasmático regulador (regulatory, R), con múltiples puntos de fosforilación para las cinasas A y C, lo que facilita que el canal sea regulado fisiológicamente por hormonas que actúan mediante dichas cinasas. En este sentido, se ha sugerido que el fármaco podría unirse al dominio citoplasmático de unión a nucleótidos (nucleotid binding domain, NBD).
En pruebas in vitro, el ivacaftor incrementó el transporte de cloruro a través de la membrana en líneas celulares con mutaciones de la CFTR (Gly551Asp, Gly1244Glu, Gly1349Asp, Gly178Arg, Gly551Ser, Ser1251Asn, Ser1255Pro, Ser549Asn o Ser549Arg; multiplicándolo por una factor que va desde 16 (Gly551Ser) a más de 1.000 (Ser549Arg).
El fármaco parece ser capaz de producir en los pacientes tratados mejoras sostenidas aunque modestas en la función respiratoria, aumento de peso corporal e incremento del porcentaje de pacientes libres de exacerbaciones pulmonares, todo ello con una reducción sustancial de la concentración de cloruro en sudor. Sin embargo, por el momento, no se ha observado una correlación entre los resultados in vitro y la respuesta del cloro en el sudor o del volumen espiratorio máximo en un segundo o VEMS (AEMPS, 2015).
Por otro lado, el mecanismo de acción y la respuesta de cloruro en sudor parecen ser independientes de la edad y debe tenerse en cuenta que puede haber enfermedad pulmonar sin que exista afectación del VEMS.
ASPECTOS MOLECULARES
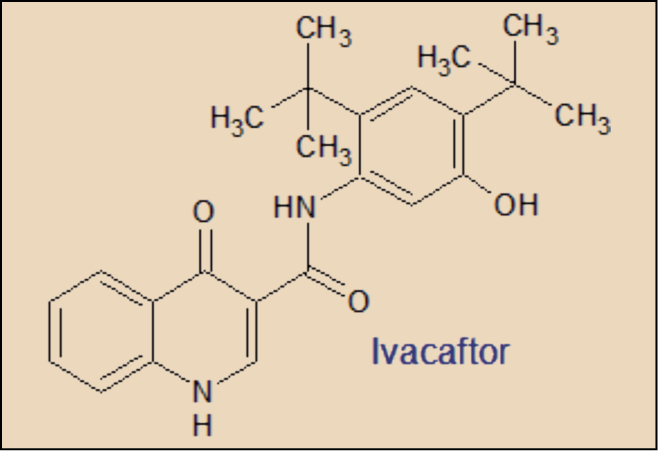
Con la mejora del conocimiento de la estructura proteica de la CFTR se ha producido el desarrollo de nuevas moléculas potencialmente útiles para mejorar el transporte de los iones cloruro en las células bronquiales de los pacientes con fibrosis quística. En general, se trata de pequeñas moléculas que pueden encuadrarse en tres grandes grupos en relación con su efecto sobre la CFTR: correctores, potenciadores y supresores. Los correctores permiten que la migración de las CFTR – tras su síntesis – hacia la membrana celular del epitelio respiratorio sea adecuada; los potenciadores permiten la activación del canal CFTR, incrementando su tiempo de apertura, mientras que los supresores impiden la finalización prematura de la síntesis de CFTR provocada por las mutaciones sin sentido que contienen codones de truncamiento.
El ivacaftor fue seleccionado dentro de una serie formada por más de 228.000 pequeñas moléculas, sintetizadas con el fin de ser testadas mediante ensayos in vitro para identificar potenciadores de CFTR. Tras la selección del ivacaftor como potencial candidato, fue estudiado en líneas celulares epiteliales bronquiales humanas, tanto primarias como recombinantes (Van Goor, 2009).
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del ivacaftor han sido adecuadamente contrastadas en las indicaciones autorizadas mediante varios ensayos clínicos de fase 3 (confirmatorios de eficacia y seguridad), aleatorizados, doblemente ciegos y controlados con placebo.
En los dos estudios realizados sobre pacientes con fibrosis quística con al menos una mutación Gly551Asp (G551D) en uno de los alelos y con un valor basal del porcentaje predicho del Volumen Espiratorio Forzado durante el primer segundo (VESM, VEF1) no inferior al 40%; el peso mínimo aceptable para los pacientes pediátricos fue de 15 kg. Ambos estudios tuvieron una duración de 48 semanas, totalizando 213 pacientes aleatorizados. Las dosis de ivacaftor fueron de 150 mg/12 h por vía oral, junto con el tratamiento que estuviesen recibiendo los pacientes. Las características demográficas, antropométricas y clínicas están especificadas en la tabla 1, junto con los principales resultados obtenidos.
|
Tabla 1. Estudios clínicos con ivacaftor: características y resultados |
|||
|
Comparador |
Placebo |
Placebo |
Placebo |
|
Indicación estudiada |
Mutación G551D |
Mutación G551D |
Mutación clase II no G551D |
|
Duración estudio |
48 semanas |
48 semanas |
8 semanas |
|
Pacientes aleatorizados |
161 |
52 |
39 |
|
Edad, mediana (rango) |
24 años >12 años (100%) |
9,0 años 6-12 años (100%) |
23 años 6-11 años (21%) 12-17 años (28%) >17 años (51%) |
|
Sexo (varones) |
48% |
48% |
56% |
|
Raza blanca/caucásica |
98% |
87% |
74% |
|
Altura basal (mediana) |
|
132 cm |
166 cm |
|
Peso basal (mediana) |
58,8 kg |
28,6 kg |
59,0 kg |
|
VESM basal (mediana) |
67% |
85% |
84% |
|
Concentración Cl– en sudor basal (mediana) |
100,5 mmol/L. |
105,0 mmol/L. |
102,0 mmol/L. |
|
Tiempo determinación de la variable principal |
24 semanas |
24 semanas |
8 semanas |
|
Variación absoluta del VESM |
10,4% IC95% 8,6 a 12,6 |
12,5% IC95% 6,6 a 18,3 |
10,7% IC95% 7,3 a 14,1 |
|
Variación absoluta del VESM (48 semanas) |
10,5% IC95% 8,5 a 12,5 |
10,0% IC95% 4,5 a 15,5 |
|
|
Variación absoluta en el dominio respiratorio |
8,1 IC95% 4,7 a 11,4 |
6,1 IC95% -1,4 a 13,5* |
9,6 IC95% 4,5 a 14,7 (8 sem.) |
|
Variación absoluta en el dominio respiratorio |
8,6 IC95% 5,3 a 11,9 |
5,1 IC95% -1,6 a 11,8* |
|
|
Variación absoluta de la concentración Cl– |
-49,7 IC95% -51,3 a -44,5 |
-54,3 IC95% -61,8 a -46,8 |
-49,6 IC95% -57,8 a -41,5 (8 sem.) |
|
Variación absoluta de la concentración Cl– |
-48,1 IC95% -51,5 a -44,7 |
-53,5 IC95% -60,9 a -46,0 |
|
|
Reducción del riesgo de exacerbación pulmonar (24 semanas) |
60% (HR=0,40; IC65% 0,23 a 0,71) |
|
|
|
Reducción del riesgo de exacerbación pulmonar (48 semanas) |
54% (HR=0,46; IC65% 0,28 a 0,73) |
|
|
|
Variación del peso corporal (24 semanas) |
2,8 kg IC95% 1,8 a 3,7 |
1,9 kg IC95% 0,9 a 2,9 |
|
|
Variación del peso corporal (48 semanas) |
2,8 kg IC95% 1,8 a 3,7 |
2,8 kg IC95% 1,3 a 4,2 |
|
|
Variación media del peso ajustado a la edad (puntuación z, a las 8 semanas) |
|
|
0,26 kg IC95% 0,12 a 0,39 |
Todas las diferencias entre los datos correspondientes al ivacaftor y al placebo fueron estadísticamente significativas, salvo aquellas notadas con un asterisco (*).
La variable primaria de eficacia utilizada consistió en la variación del VESM a las 24 semanas; como variables secundarias se determinó este mismo parámetro a las 48 semanas (como indicador de la durabilidad de la respuesta), así como la variación absoluta de la puntuación del dominio respiratorio del Cuestionario revisado para fibrosis quística (Cystic Fibrosis Questionnaire Revised, CFQ-R), una medida de los síntomas respiratorios relevantes para los pacientes con fibrosis quística, tales como tos, producción de esputo y dificultad para respirar. También se determinó la variación de la concentración de cloruro en el sudor, del peso corporal (y de la altura, en el caso de los pacientes pediátricos) y el efecto sobre el riesgo de experimentar exacerbaciones pulmonares (en los pacientes adultos).
Por lo que se refiere al estudio aleatorizado, doblemente ciego, cruzado y controlado con placebo, realizado sobre pacientes con fibrosis quística con al menos una mutación de clase III que no sea la Gly551Asp (G551D)5, tuvo una duración de 8 semanas (con cada tratamiento), totalizando 39 pacientes. Las dosis de ivacaftor fueron de 150 mg/12 h por vía oral, junto con el tratamiento que estuviesen recibiendo los pacientes. Las características demográficas, antropométricas y clínicas están especificadas en la tabla 1, junto con los principales resultados obtenidos. La variable primaria de eficacia fue idéntica a los anteriores estudios, aunque determinada a las 8 semanas.
Desde el punto de vista de la seguridad, el ivacaftor presenta un perfil toxicológico relativamente benigno, con una incidencia general de eventos adversos similar a la registrada con placebo. En los estudios clínicos controlados, los eventos adversos descritos con más frecuencia que con placebo fueron cefalea (16,7% vs. 14,4% con placebo), infecciones del tracto respiratorio superior (15,8 vs. 12,1%), congestión nasal (15,8 vs. 12,9%), erupciones cutáneas (10,4 vs. 5,3%), rinitis (5,9 vs. 3,0%) y vértigo (5,4 vs. 2,3%).
Aunque algunos estudios han reportado la aparición de incrementos notables de los niveles de enzimas hepáticos, particularmente de transaminasas (ALT y AST), es preciso tener en cuenta que tales elevaciones son comunes en los pacientes con fibrosis quística. De hecho, en los estudios controlados con placebo, la incidencia de estas elevaciones fueron más frecuentes entre los pacientes tratados con placebo que con ivacaftor (7,6 vs. 3,6%). Sin embargo, en el subgrupo de pacientes con un historial médico de elevación de transaminasas, el aumento de ALT o AST se han reportado con mayor frecuencia en los pacientes que recibieron ivacaftor en comparación con el placebo. Por este motivo, se recomiendan las pruebas de función hepática en todos los pacientes antes de iniciar ivacaftor, así como cada 3 meses durante el primer año de tratamiento, y posteriormente cada año. Para todos los pacientes con antecedentes de elevaciones de transaminasas, debería considerarse un seguimiento más frecuente de las pruebas de función hepática. En cualquier caso, los pacientes que presenten un aumento de las transaminasas deben ser estrechamente monitorizados hasta que las anomalías se resuelvan y la administración de ivacaftor debe interrumpirse en pacientes con niveles de ALT o AST que superen en más de 5 veces el límite superior de la normalidad.
ASPECTOS INNOVADORES
El ivacaftor es un potenciador de la proteína CFTR, es decir, incrementa la actividad del canal de cloruro (CFTR) debido a un aumento del tiempo durante el cual el canal permanece abierto. Ha sido autorizado como medicamento huérfano para el tratamiento de pacientes con fibrosis quística de 6 años de edad y mayores con una de las siguientes mutaciones de apertura del canal (clase III) en el gen CFTR: Gly551Asp (G551D), Gly1244Glu (G1244E), Gly1349Asp (G1349D), Gly178Arg (G178R), Gly551Ser (G551S), Ser1251Asn (S1251N), Ser1255Pro (S1255P), Ser549Asn (S549N) o Ser549Arg (S549R). La proteína CFTR es un canal de cloruro (Cl–) regulado por AMPc, que se localiza en la membrana de las células epiteliales, donde actúa regulando el flujo de electrolitos y de agua. Su disfunción – de origen genético – modifica la cantidad y la composición relativa de los fluidos, afectando a los órganos con tejido epitelial, provocando una excesiva absorción de fluidos y la correspondiente deshidratación de la superficie epitelial, que es responsable de la formación de un moco hiperviscoso (mucoviscidosis) con un aumento de la concentración mucina polimérica, característica de la fibrosis quística y responsable de la mayor parte de los trastornos clínicos ligados a la enfermedad.
Los datos clínicos disponibles son limitados aunque apuntan claramente a una mejoría de la función respiratoria que, aunque modesta (incremento del volumen espiratorio forzado durante el primer segundo o VESM, en torno al 10-13%), es estadística significativa y clínicamente relevante; en cualquier caso, es superior al conseguido con los tratamientos utilizados actualmente (AEMPS, 2015). Un aspecto interesante es que la respuesta al tratamiento con ivacaftor se mantuvo constante durante al menos un año, tanto en adultos como en niños (≥6 años) con la mutación Gly551Asp (G551D). Otras variables utilizadas, como las variaciones de la puntuación del dominio respiratorio del cuestionario revisado para fibrosis quística (CFQ-R), la de la concentración de cloruro en el sudor y la del peso corporal (y de la altura, en el caso de los pacientes pediátricos), así como la reducción del riesgo de experimentar exacerbaciones pulmonares (en los pacientes adultos), complementan un perfil terapéutico favorable.
Asimismo, el ivacaftor presenta un perfil toxicológico relativamente benigno, con una incidencia general de eventos adversos similar a la registrada con placebo. En los estudios clínicos controlados, los eventos adversos descritos con más frecuencia que con placebo fueron cefalea, infecciones del tracto respiratorio superior, congestión nasal, erupciones cutáneas, rinitis y vértigo.
A pesar de estos perfiles terapéuticos y toxicológicos moderadamente satisfactorios, conviene recordar que el tratamiento no es curativo y, por tanto, sería preciso mantener de forma crónica el tratamiento a lo largo de la vida (o, al menos, durante periodos prolongados), lo cual plantea la duda sobre la durabilidad de la eficacia y de la seguridad a largo plazo (aunque los datos a un año sugieren estabilidad en la respuesta). Por otro lado, los datos clínicos más consistentes – siempre relativamente – corresponden a pacientes con fibrosis quística con la mutación Gly551Asp (G551D), mientras que los correspondientes a otras mutaciones de clase III son mucho más limitados. Por otro lado, existen numerosas lagunas de información sobre la eficacia y la seguridad en determinadas poblaciones de pacientes, particularmente en menores de 6 años, embarazadas, con insuficiencia hepática grave, bajo tratamiento con fármacos inhibidores del citocromo CYP3A4 (implicado en el metabolismo hepático del ivacaftor) o trasplantados.
Aunque la fisiopatología de la enfermedad es similar en adultos y población pediátrica, las manifestaciones clínicas dependen en gran medida de la edad. En general, el deterioro de la función respiratoria es menor en niños y en adolescentes que en adultos, mientras que el mecanismo de acción del fármaco y la respuesta relativa a la concentración de cloruro en sudor son independientes de la edad, ya que la glándula sudorípara no se desestructura con el tiempo y la enfermedad. Por otro lado, puede haber enfermedad pulmonar sin que exista afectación del VEMS; en este sentido, la enfermedad pulmonar es aparente en un 40-50% de niños y adolescentes mientras que menos del 20% de estos pacientes presentan un VEMS ≤90%.
Según el Informe de Posicionamiento Terapéutico de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS, 2015), la decisión de iniciar tratamiento con ivacaftor deberá tomarse de manera individual, considerando la situación clínica de cada paciente, así como el tratamiento que esté recibiendo, y que generalmente incluye fisioterapia respiratoria, dornasa alfa, suero salino hipertónico, antibióticos inhalados, etcétera. Asimismo, en aquellos pacientes que inicien tratamiento con ivacaftor, la necesidad de mantener el mismo deberá ser reevaluada periódicamente en base a su efectividad.
Sin duda, el ivacaftor representa un avance esperanzador en la lucha contra la fibrosis quística, al ser el primer medicamento que actúa sobre uno de los componentes biomoleculares ligados etiológicamente a la enfermedad. Sin embargo, su utilidad está limitada a la fibrosis quística en pacientes con mutaciones de apertura del canal (clase III) en el gen CFTR, que representan menos del 5% de todos los casos. Por el contrario, el ivacaftor ha demostrado ser ineficaz en los cuadros ligados a mutaciones de clase II, en particular la Phe508del (F508del) responsables de al menos un 50% de los casos de fibrosis quística. Esto implica que el ivacaftor es lo que se conoce como un medicamento ultra-huérfano, en tanto que las variantes genéticas de la fibrosis quística incluidas en las indicaciones autorizadas suponen una enfermedad ultra-rara, considerando como tal a aquella que afecta a menos de 40.000 pacientes en toda la Unión Europea.
Con todo, se trata, sin duda, de una innovación interesante.
|
VALORACIÓN |
|
|
IVACAFTOR
|
|
|
Grupo Terapéutico (ATC): R07AX. APARATO RESPIRATORIO. Otros preparados. |
|
|
Indicaciones autorizadas: tratamiento de pacientes con fibrosis quística (FQ) de 6 años de edad y mayores con una de las siguientes mutaciones de apertura del canal (clase III) en el gen CFTR: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N o S549R |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar. |
♣ ♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar. Supone la incorporación de una nueva vía terapéutica en ausencia de alternativas terapéuticas. Posibilidad de asociar con otros tratamientos actualmente en vigor. |
⇑ |
|
Novedad molecular: Mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma o similar indicación terapéutica. |
⇑ |

|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Ivacaftor |
Kalydeco |
Vertex |
2015 |
BIBLIOGRAFÍA
Bibliografía
- Agencia Española de Medicamentos y Productos Sanitarios. Informe de Posicionamiento Terapéutico de ivacaftor (Kalydeco®). 17 de junio de 2015. http://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-ivakaftor-kalydeco-aprobado-GCPT.pdf
- Alonso MJ, Heine-Suñer D, Calvo M, Rosell J, Giménez J, Ramos MD, et al. Spectrum of mutations in the CFTR gene in cystic fibrosis patients of Spanish ancestry. Ann Hum Genet. 2007; 71(Pt 2): 194-201.
- Bellón G. Fibrosis quística. www.orpha.net (2006)
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Fibrosis quística. Panorama Actual Med. 2014, 38(376): 711-21.
- Döring G, Flume P, Heijerman H, Elborn JS; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J Cyst Fibros. 2012; 11: 461-79. doi:10.1016/j.jcf.2012.10.004
- Ehre C, Ridley C, Thornton DJ. Cystic fibrosis: An inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol. 2014; 52C: 136-45. doi: 10.1016/j.biocel.2014.03.011.
- European Medicines Agency (EMA). Kalydeco. Assessment report for paediatric studies submitted according to Article 46 of the Regulation (EC) No 1901/2006. 26 February 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/002494/WC500185166.pdf
- European Medicines Agency (EMA). Kalydeco. Assessment report. EMA/523198/2014. 26 June 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002494/WC500130766.pdf
- European Medicines Agency (EMA). Kalydeco. Assessment report. EMA/473279/2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002494/WC500130766.pdf
- Federación Española de Fibrosis Quística (FEGQ). http://fibrosisquistica.org
- Lerín M, Prados C, Martínez MT, Maíz L, Girón R, Solé A, Cabanillas JJ, Alvarez-Sala R. Cystic fibrosis in adult age. Rev Clin Esp. 2014 Jun 16. pii: S0014-2565(14)00209-4. doi: 10.1016/j.rce.2014.05.001.
- Ministerio de Sanidad y Consumo (MSC). Libro blanco de la atención a la fibrosis quística (2010). http://www.fibrosisquistica.org/images/recursos/31.pdf
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al; VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011; 365(18): 1663-72. doi: 10.1056/NEJMoa1105185.
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009; 106(44): 18825-30. doi: 10.1073/pnas.0904709106.
1 En la Unión Europea, el criterio demográfico para calificar como enfermedad rara es una incidencia de 5 casos por cada 10.000 habitantes (1:2.000).
2 Hasta un 30% en los recién nacidos que tienen un hermano que a su vez tuvo íleo meconial.
3 El vólvulo es un giro del intestino sobre sí mismo. Es una de las causas más comunes de la obstrucción intestinal, especialmente en niños.
4 La notación actual de las mutaciones puntuales (que implican a un único aminoácido en la proteína sintetizada) indican la sustitución del aminoácido citado en primer lugar por el indicado en segundo lugar; la eliminación del aminoácido (deleción) pero sin truncamiento de la proteína, se nota con el término del; finalmente, cuando la mutación implica el truncamiento de la proteína en el punto designado, se especifica con una X al final. Anteriormente, los aminoácidos eran designados con una única letra (estándar internacional), aunque actualmente se opta generalmente por indicar los aminoácidos con su notación estándar internacional de tres letras.
5 Concretamente: Gly1244Glu (G1244E), Gly1349Asp (G1349D), Gly178Arg (G178R), Gly551Ser (G551S), Ser1251Asn (S1251N), Ser1255Pro (S1255P), Ser549Asn (S549N) o Ser549Arg (S549R).
Artículos relacionados
-
5 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
29 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
6 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
