Eliglustat ▼(Sanofi Aventis) en enfermedad de Gaucher
 Nº400
Nº400

Resumen
El eliglustat es un inhibidor selectivo de la glucosilceramida sintasa (UDP-ceramida sintasa), que cataliza la biosíntesis de glucoesfingolípidos a partir de UDP-glucosa y ceramida, lo que permite reducir la acumulación de glucofsilceramida dentro de los lisosomas, ante un déficit o disfunción bioquímica de la b-glucosidasa ácida (b-glucocerebrosidasa) natural, tal como ocurre en la enfermedad de Gaucher. El medicamento (huérfano) ha sido autorizado para el tratamiento a largo plazo de pacientes adultos con enfermedad de Gaucher de tipo 1. Los datos procedentes de los estudios clínicos de referencia sugieren un efecto clínicamente relevante, con reducción significativa de la esplenomegalia y la hepatomegalia, y mejoras en los parámetros hematológicos. La sustitución de la terapia enzimática de restauración por eliglustat no parece afectar negativamente a los pacientes y, en cambio, mejora su calidad de vida al sustituir la administración IV cada dos semanas por una cómoda posología oral. Desde el punto de vista de la seguridad, el eliglustat presenta un perfil toxicológico relativamente benigno, asociándose a eventos adversos generalmente leves y transitorios (cefalea, artralgia, nasofaringitis, infección del tracto respiratorio superior, diarrea y vértigo), sustancialmente mejores que con su competidor, el miglustat. Todo ello sugiere un posible papel para el eliglustat como tratamiento de primera línea para pacientes con enfermedad de Gaucher de tipo 1 que no hayan mejorado sustancialmente con la terapia de restauración enzimática.
ASPECTOS FISIOPATOLÓGICOS
Se distinguen dos tipos básicos de metabolopatías que afectan a las proteínas intracelulares:
- Síntesis defectuosa o ausencia total de proteínas estructurales. Dentro de este apartado se incluye a un amplio grupo de anemias hemolíticas congénitas.
- Almacenamiento anómalo en lisosomas. Una de las misiones esenciales que tienen los lisosomas (pequeños orgánulos intracelulares ricos en enzimas de muy diverso tipo) es degradar muchas de las sustancias producidas por la propia célula. De ahí, que la deficiencia de un enzima específico provoque la acumulación del correspondiente sustrato, potencialmente lesivo para la célula y, en última instancia, para el conjunto del organismo.
Las enfermedades por almacenamiento anómalo en lisosomas constituyen una amplia variedad de deficiencias enzimáticas que provocan la acumulación de diversas sustancias en el interior de los lisosomas: glucoproteínas, mucopolisacáridos, glucolípidos, oligosacáridos, etc. Los síntomas varían según el tipo de producto acumulado, pero la mayoría conduce a graves alteraciones neurológicas y la evolución suele ser fatal durante los primeros años de vida; generalmente, tienen un carácter hereditario de tipo autosómico recesivo. Un ejemplo de éstas es la enfermedad de Gaucher, que es debida a una alteración del almacenamiento de lípidos. Fue descrita por primera vez por Philippe Gaucher en 1882, identificándose su causa como la deficiencia o ausencia de actividad de la enzima β-glucosidasa, también conocida como β-glucocerebrosidasa o glucosilceramidasa EC3.2.1.45 (GBA1), que conduce a la acumulación de glucocerebrósido o glucosilceramida (GLC).
La enfermedad parece afectar a hombres y mujeres por igual y se manifiesta bajo tres presentaciones clínicas: la de tipo 1 (no neuronopática), la de tipo 2 (neuronopática aguda) y la de tipo 3 (neuronopática crónica), en función de la presencia o no de deterioro neurológico, la edad en la identificación y el grado de progresión de la enfermedad. Los pacientes con los tipos 2 y 3 manifiestan complicaciones más graves que aquellos con la de tipo 1, muriendo muchos de ellos a edades tempranas (incluso infantiles). Por el contrario, la enfermedad de Gaucher de tipo 1 ocurre principalmente en adultos y representa el 95% de los casos; en aquellos casos en los que la manifestación ocurre antes de la edad adulta, es probable que tenga una progresión más rápida.
Su incidencia en la población general es de 1:20.000-200.000 habitantes, aunque entre poblaciones intensamente endogámicas como la de los judíos Ashkenazi, son portadores de genes defectuosos entre el 8% y el 10% de la población, la incidencia es muchos más común (1:400-1.000). En España, la incidencia general es muy baja, estimándose del orden de 1:200.000, aunque la alta proporción de sujetos asintomáticos (entre un 10% y un 15% de los casos) sugieren una incidencia real mayor de la observada.
Bioquímicamente, la enfermedad de Gaucher se caracteriza por la acumulación de glucosilceramida (glucocerebrósido), como consecuencia de un déficit o una deficiencia funcional de ß-glucosidasa ácida (ß-glucocerebrosidasa). La glucosilceramida es un intermedio normal en el catabolismo de globósidos y gangliósidos, y su hidrólisis a ceramida y glucosa está catalizada por el enzima indicado. El glucocerebrósido deriva principalmente del recambio celular hematopoyético.
La acumulación del lípido glucocerebrósido en los macrófagos-monocitos tisulares da lugar a las denominadas células de Gaucher. Estas células se caracterizan por tener un diámetro de 20-100 μm, pequeños núcleos excéntricos y citoplasma con arrugas y estriación; se encuentran principalmente en hígado, bazo y médula ósea, apareciendo de forma esporádica en pulmón, riñones e intestino, causando secuelas hematológicas (anemia y trombocitopenia graves), esplenomegalia progresiva y complicaciones esqueléticas muy comunes e incapacitantes (osteonecrosis, osteopenia, fracturas patológicas secundarias, etc.). La acumulación de células de Gaucher incrementa la producción de citocinas proinflamatorias, que son responsables del agrandamiento del bazo y del hígado, la destrucción ósea, las anomalías pulmonares, la anemia, la trombopenia y la leucopenia.
La disminución en la actividad de la enzima glucocerebrosidasa en la enfermedad de Gaucher está causada por mutaciones en el gen de la glucocerebrosidasa o en el de su activador, saposina C. Se han descrito unas 60 mutaciones del gen de la glucocerebrosidasa, y al menos dos del de la saposina C. Las mutaciones más frecuentemente encontradas en el gen de la glucocerebrosidasa, con independencia del origen étnico de los individuos, corresponden a las sustituciones de asparagina por serina en el aminoácido 370 (N370S) y leucina por prolina en la posición 444 (L444P).
No existe un tratamiento definitivo actualmente para este tipo de enzimopatías, aunque son candidatas claras, por su origen monogénico, al desarrollo de terapias génicas específicas. Afortunadamente, se han desarrollado tratamientos de restauración enzimática para algunas de ellas, consistentes en la administración exógena del enzima deficitario o disfuncional, actualmente de origen recombinante en la gran mayoría de los casos. En el caso de la enfermedad de Gaucher se dispone desde 1999 de la forma recombinante del enzima, imiglucerasa (Cerezyme®) y anteriormente estuvo comercializada la alglucerasa (Ceredase®) hasta que fue sustituida por la imiglucerasa; en 2011 se comercializó la velaglucerasa alfa (Vpriv®) y, posteriormente, la taliglucerasa alfa (Elelyso®), aunque esta última no está comercializada en España.
La terapia de restauración enzimática es especialmente eficaz en los trastornos hematológicos (mejoría de la anemia en un 30% y la trombopenia en un 40%) y el tratamiento de las visceromegalias (la hepatomegalia se reduce un 20% y la esplenomegalia un 35%), con evidentes resultados ya en los primeros 6 meses del tratamiento. Las manifestaciones óseas, aunque de modo más lento, también responden satisfactoriamente a la infusión de la enzima. Además, la terapia restauración enzimática posee un excelente perfil de seguridad.
Otra opción terapéutica consiste en el empleo de pequeñas moléculas capaces de inhibir la glucosilceramida sintasa (UDP-ceramida sintasa), que cataliza la biosíntesis de glucoesfingolípidos a partir de UDP-glucosa y ceramida, lo que permite reducir la acumulación de glucosilceramida dentro de los lisosomas. El primer fármaco de este grupo fue el miglustat (Zavesca®), comercializado en 2004; sin embargo, debido a sus características moleculares, el miglustat también es capaz de inhibir en mayor o menor grado los enzimas α-glucosidasa I y II, implicadas en los procesos de plegamiento y funcionamiento de las proteínas; además, también inhibe a sucrasa y maltasa (dos disacaridasas), todo lo cual posiblemente se relaciona con una elevada incidencia de reacciones adversas con miglustat, como temblor (30%), diarrea (85%), pérdida de peso (65%), trombopenia, entumecimiento y sensación de ardor en las manos y los pies. Debido los papeles esenciales de los glucoesfingolípidos, la reducción a largo plazo de los niveles de estos lípidos puede afectar a una amplia variedad de funciones celulares normales. El miglustat se emplea para los pacientes que no pueden recibir la terapia de restauración enzimática debido a que hayan experimentado previamente reacciones anafilácticas a la administración exógena del enzima recombinante. Pero su baja tolerabilidad condujo al desarrollo del eliglustat (Cerdelga®), más selectivo que su antecesor y, por ello, con un mejor perfil de seguridad. Tanto miglustat como eliglustat son cómodamente administrados por vía oral, a diferencia de los enzimas de restauración, que requieren administración IV.
ACCIÓN Y MECANISMO
El eliglustat es un inhibidor selectivo de la glucosilceramida sintasa (UDP-ceramida sintasa), que cataliza la biosíntesis de glucoesfingolípidos a partir de UDP-glucosa y ceramida, lo que permite reducir la acumulación de glucosilceramida dentro de los lisosomas, ante un déficit o disfunción bioquímica de la ß-glucosidasa ácida (ß-glucocerebrosidasa) natural, tal como ocurre en la enfermedad de Gaucher. El medicamento ha sido autorizado para el tratamiento a largo plazo de pacientes adultos con enfermedad de Gaucher de tipo 1 (EG1).
Biológicamente, los glucoesfingolípidos participan en los procesos de crecimiento y diferenciación celular, organización de la membrana celular y señalización bioquímica. Además de ser un elemento estructural esencial para la síntesis de esfingolípidos (glucoesfingolípidos y esfingomielinas), es también una potente molécula de señalización. Por su parte, la glucosilceramida parece tener un importante papel en el mantenimiento del crecimiento de los axones neuronales. Es, pues, es un intermedio normal en el catabolismo de globósidos y gangliósidos, y su hidrólisis a ceramida y glucosa está catalizada por la ß-glucosidasa ácida (ß-glucocerebrosidasa), deficiente o disfuncional en la enfermedad de Gaucher.
ASPECTOS MOLECULARES
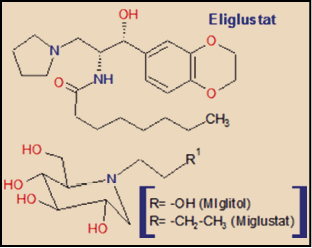
El eliglustat guarda una cierta familiaridad química con el miglustat, un iminoazúcar que actúa como inhibidor de la glucosilceramida sintasa (glucosiltransferasa), así como de los enzimas α-glucosidasa I y II, y de sucrasa y maltasa (dos disacaridasas) y que deriva de la 1-desoxinojirimicina, por lo que también está estrechamente relacionado con el miglitol, un inhibidor de α-glucosidasas presentes en la superficie interna del intestino delgado, utilizado como agente hipoglucemiante (reduce la conversión de sacarosa en glucosa y fructosa), pero responsable también de efectos adversos gastrointestinales y diarrea osmótica, fruto de sus efectos sobre el metabolismo glucídico intestinal.
Existen algunas importantes diferencias estructurales entre eliglustat y miglustat. La más relevante es que la molécula de eliglustat recuerda a la fracción de ceramida de la glucosilceramida, mientras que el miglustat está más próximo estructuralmente a la fracción de glucosa de la glucosilceramida, lo que determina una mayor selectividad del eliglustat sobre la glucosilceramida sintasa; por otro lado, esta misma diferencia estructural determina que el eliglustat carezca de efectos significativos sobre los enzimas α-glucosidasa I y II, y sobre las disacaridasas sucrasa y maltasa, con lo que la incidencia de efectos adversos digestivos es notablemente inferior que con el miglustat.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del eliglustat han sido adecuadamente contrastadas en la indicación autorizada mediante dos ensayos clínicos de fase 3 (confirmatorios de eficacia y seguridad), aleatorizados, multicéntricos y multinacionales. El primero de ellos (ENGAGE; Mistry, 2015) fue doblemente ciego, controlado con placebo y desarrollado sobre un total de 40 pacientes no tratados anteriormente en 18 centros de 12 países; el segundo fue un estudio abierto de no inferioridad (ENCORE; Cox, 2015), en el que se comparó el eliglustat con el tratamiento de restauración enzimática (imiglucerasa) en 160 pacientes procedentes de 39 centros de cuatro continentes.
En el primero de los estudios (ENGAGE) se incluyó a 40 pacientes adultos con enfermedad de Gaucher de tipo 1 con esplenomegalia más trombocitopenia o anemia, que no habían sido tratados específicamente con anterioridad (naïve). Los pacientes fueron estratificados en función del grado de esplenomegalia y fueron asignados aleatoriamente a recibir por vía oral eliglustat (50 o 100 mg/12 h) o placebo durante un periodo de nueve meses. La variable primaria de eficacia consistió en el grado de variación del volumen del bazo (expresado múltiplos del tamaño normal) durante los nueve meses de tratamiento; como variable secundaria se determinó la variación en el nivel de hemoglobina y los cambios porcentuales en el volumen del hígado y del recuento plaquetario.
Los resultados mostraron una reducción media del volumen del bazo del 27,8% (IC95% -32,6 a -23,0; de 13,9 a 10,2 múltiplos del tamaño normal del bazo) en el grupo tratado con eliglustat frente un incremento medio del 2,3% (IC95% -2,5 a 7,1; de 12,5 a 12,8 múltiplos del bazo normal) con placebo, lo que supone una diferencia absoluta entre ambos tratamientos del 30,0 puntos porcentuales (IC95% -36,8 a -23,2; p<0,001). En cuanto a las variables secundarias, las diferencias absolutas favorecieron claramente al grupo tratado con eliglustat, con una diferencias medias absolutas de +1,22 g/dl de incremento de los niveles de hemoglobina (IC95% 0,57 a 1,88; p<0,001), una reducción del 5,5% del volumen hepático (IC95% -11,4 a -1,9; p=0,007) y un incremento del 41,1% en el recuento de plaquetas (IC95% 24,0 a 58,2; p<0,001).
En el segundo de los estudios (ENCORE) se seleccionó a 160 pacientes adultos con enfermedad de Gaucher de tipo 1 que había recibido terapia de restauración enzimática con imiglucerasa (infusión IV), asignándoles aleatoriamente para mantener dicho tratamiento o sustituirlo por eliglustat (100-150 mg/12 h, oral) durante un periodo de un año. La variable clínica principal fue el porcentaje de pacientes que estabilizaron su condición, cumpliendo con un criterio compuesto formado por parámetros bioquímicos y de volumen orgánico al final del periodo (niveles de hemoglobina no inferiores a 15 g/l, reducción del recuento plaquetario no superior del 25%, incremento no superior al 25% en el volumen del bazo e incremento no superior al 20% en el volumen del hígado). Se consideró como margen de inferioridad un 25% para el grupo tratado con eliglustat, con relación al de imiglucerasa.
Los resultados mostraron que en la población sujeta a protocolo, la tasa de pacientes que mantuvieron su condición clínica estable (variable primaria compuesta) fue del 85% con eliglustat vs. 94% con imiglucerasa, siendo la diferencia entre grupos de -8,8 puntos porcentuales (IC95% -17,6 a +4,2), estadísticamente no significativa.
Desde el punto de vista de la seguridad, el eliglustat presenta un perfil toxicológico relativamente benigno, asociándose a eventos adversos generalmente leves y transitorios. Los más comunes que aparecieron durante el tratamiento fueron cefalea (17%), artralgia (14%), nasofaringitis (13%), infección del tracto respiratorio superior (11%), diarrea (10%) y vértigo (10%).
La frecuencia de eventos adversos intensos (severos) emergentes durante el tratamiento fue del 9,0%, que estaban mayoritariamente ligados a la hospitalización por la aparición de otras patologías graves que pudieran incrementar el riesgo para los pacientes con enfermedad de Gaucher. Entre ellos, los más frecuentemente descritos fueron infarto de miocardio (4 pacientes) y síncope (5 pacientes), sobre un total de 393 pacientes controlados. Un 3% de los pacientes suspendió el tratamiento con eliglustat debido a la aparición de eventos adversos durante el tratamiento.
ASPECTOS INNOVADORES
El eliglustat es un inhibidor selectivo de la glucosilceramida sintasa (UDP-ceramida sintasa), que cataliza la biosíntesis de glucoesfingolípidos a partir de UDP-glucosa y ceramida, lo que permite reducir la acumulación de glucosilceramida dentro de los lisosomas, ante un déficit o disfunción bioquímica de la ß-glucosidasa ácida (ß-glucocerebrosidasa) natural, tal como ocurre en la enfermedad de Gaucher. El medicamento (huérfano) ha sido autorizado para el tratamiento a largo plazo de pacientes adultos con enfermedad de Gaucher de tipo 1 (EG1).
Los datos registrados en los estudios clínicos de referencia sugieren un efecto clínicamente relevante. En concreto, en el estudio ENGAGE, tras nueve meses de tratamiento con eliglustat el volumen medio del bazo fue reducido en un 30% y el del hígado en un 6%, mientras que aumentaban los niveles de hemoglobina en 1,22 g/l y el recuento plaquetario en un 41%, encontrándose los mejores resultados en los pacientes más gravemente afectados por la enfermedad de Gaucher. Por otro lado, los datos del estudio ENCORE han mostrado la no inferioridad del eliglustat durante un año con respecto a la terapia estándar de restauración enzimática, en el mantenimiento de los parámetros hematológicos y de los volúmenes orgánicos (bazo e hígado) en pacientes tratados previamente durante al menos tres años con dicha terapia de restauración.
Desde el punto de vista de la seguridad, el eliglustat presenta un perfil toxicológico relativamente benigno, asociándose a eventos adversos generalmente leves y transitorios. Los más comunes (10-20%) son cefalea, artralgia, nasofaringitis, infección del tracto respiratorio superior, diarrea y vértigo. Se ha observado que con dosis varias veces superiores a las utilizadas en clínica, el eliglustat se asociaba con un alargamiento del intervalo QT del electrocardiograma, aunque no han observado anomalías cardiovasculares asociadas con las dosis utilizadas en los dos ensayos clínicos de referencia. No obstante, ante la ausencia de experiencia clínica en pacientes con patología cardíaca previa, se contraindica su uso en estos pacientes y, con el de evitar elevadas concentraciones plasmáticas del medicamento, se recomienda una dosificación personalizada en función de la capacidad metabolizadora, determinando el estatus del genotipo CYP2D6 por ser este isoenzima del citocromo P450 el principal responsable de su metabolismo hepático.
La aparición de la terapia de restauración enzimática para la enfermedad de Gaucher en la década de los 90 del siglo pasado supuso una notable mejora la situación clínica de estos pacientes y, de hecho, se han convertido en la terapia estándar para estos pacientes; sin embargo, la terapia de restauración enzimática con imiglucerasa o velaglucerasa alfa tiene algunas limitaciones importantes. La primera de estas limitaciones es la necesidad de administrarla en forma de infusión IV lo, dado el carácter no curativo del tratamiento, obliga a esta forma de administración de forma persistente durante toda la vida; obviamente, la vía IV, en especial cuando se requiere utilizar cada dos semanas (como es el caso de imiglucerasa y velaglucerasa alfa), afecta de forma significativa a la calidad de vida de los pacientes.
Por otro lado, aunque estos enzimas tienen un perfil de seguridad aceptable, mantienen el riesgo inevitable de reacciones alérgicas durante las infusiones IV, lo que contraindica posteriores administraciones o producen anticuerpos que pueden reducir la eficacia a largo plazo. Por otro lado, al tratarse de enzimas y, por tanto, tener un elevado peso molecular, no son susceptibles de atravesar la barrera hematoencefálica y con ello no podrían utilizarse en los tipos 2 y 3 (neuronopáticas) de la enfermedad de Gaucher. No obstante, ni el miglustat ni el eliglustat han sido estudiados específicamente en estos tipos por el momento, al menos en ensayos clínicos controlados.
No hay estudios directamente comparativos entre eliglustat y miglustat. Sin embargo, de forma indirecta se manifiesta una evidente diferencia en cuanto a perfiles de seguridad, ya que mientras que con el eliglustat la incidencia de los más comunes no supera el 20% y son de carácter leve y transitorio, los del miglustat son mucho más comunes (86% diarrea 86%, 70% pérdida de peso, 30% temblor y casos aislados de disfunción cognitiva y neuropatía periférica, posiblemente relacionados con deficiencia de vitamina B12) y obligan a evaluaciones periódicas de las funciones neurológicas.
Los resultados de los estudios clínicos sugieren un posible papel para el eliglustat como tratamiento de primera línea para pacientes con enfermedad de Gaucher de tipo 1 que no hayan mejorado sustancialmente con la terapia de restauración enzimática (imiglucerasa, velaglucerasa alfa), con la clara ventaja de la administración oral del eliglustat, preferida por el 94% de los pacientes (Sachi, 2016).
|
VALORACIÓN |
|
ELIGLUSTAT
|
|
Grupo Terapéutico (ATC): A16AX. TRACTO ALIMENTARIO Y METABOLISMO. Otros productos |
|
Indicaciones autorizadas: Tratamiento a largo plazo de pacientes adultos con enfermedad de Gaucher de tipo 1 (EG1) que son metabolizadores lentos, metabolizadores intermedios (MI) o metabolizadores rápidos (MR) del CYP2D6. |
|
INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |

|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Miglustat |
Zavesca |
Actelion |
2004 |
|
Eliglustat |
Cerdelga |
Sanofi Aventis |
2017 |
BIBLIOGRAFÍA
Bibliografía
- Bennett LL, Turcotte K. Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Des Devel Ther. 2015; 9: 4639-47. doi: 10.2147/DDDT.S77760.
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cox TM, Drelichman G, Cravo R, Balwani M, Burrow TA, Martins AM, et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet. 2015; 385(9985): 2355-62. doi: 10.1016/S0140-6736(14)61841-9.
- Cuéllar Rodríguez S. Miglustat (Zavesca®) en enfermedad de Gaucher. Panorama Actual Med 2004; 28(272): 241-4.
- Cuéllar Rodríguez S. Velaglucerasa alfa (Vpriv®) en enfermedad de Gaucher de tipo I. Panorama Actual Med 2011; 35(343): 407-11.
- European Medicines Agency (EMA). Cerdelga®. European Public Assessment Report (EPAR). EMA/722663/2014; EMEA/H/C/003724. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003724/WC500182389.pdf
- Mistry PK, Lukina E, Ben Turkia H, Amato D, Baris H, Dasouki M, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015; 313(7): 695-706. doi: 10.1001/jama.2015.459.
- Sechi A, Dardis A, Bembi B. Profile of eliglustat tartrate in the management of Gaucher disease. Ther Clin Risk Manag. 2016; 12: 53-8. doi: 10.2147/TCRM.S73226.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
