Efmoroctocog alfa (▼Elocta®, Swedish Orphan Viovitrum) en hemofilia A
 Nº398
Nº398

Resumen
El efmoroctocog alfa es una proteína de fusión recombinante con una elevada semivida de eliminación, formada por el factor VIII de coagulación humano con deleción del dominio B, unido al dominio Fc de la inmunoglobulina humana G1 (IgG1). Ha sido autorizada para el tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A. Los datos acumulados de los dos estudios de fase III disponibles y su estudio de extensión, que todavía está en curso, han demostrado la eficacia a largo plazo de efmoroctocog alfa en el tratamiento de episodios agudos de sangrado, el manejo perioperatorio y la profilaxis en varones con hemofilia A grave tratados previamente. La sustitución del factor VIII por efmoroctocog alfa parece ser bien tolerado en pacientes previamente tratados, sin que se evidence ningún de aumento de la inmunogenicidad. Por su parte, la incidencia de eventos adversos asociados al medicamento es mínima, habiéndose documentado algunos cuadros leves de malestar, cefalea, artralgia y mialgia, descritos en un 0,2% de los pacientes. Y, lo que es de especial importancia, no se ha descrito prácticamente ningún caso de aparición de anticuerpos inhibidores. Por todo ello, el efmoroctocog alfa puede considerarse como una alternativa eficaz a las preparaciones convencionales de factor VIII, incluyendo las formas recombinantes para el tratamiento de la hemofilia A, ofreciendo una protección prolongada frente a los episodios de sangrado mediante inyecciones profilácticas cada 3 a 5 días.
ASPECTOS FISIOPATOLÓGICOS
La coagulación es un proceso extremadamente complejo que incluye una serie de reacciones generadas en la superficie de las células, que expresan en su superficie el factor tisular (FT), y cuyo objetivo es la formación de trombina en sitios de lesión vascular. La denominada de cascada de la coagulación considera un proceso de 3 fases: inicio, amplificación y acción de la trombina. La fase de inicio se produce tras la lesión vascular, cuando el factor VII (proconvertina) se une a las células que presentan en su superficie el factor tisular de tromboplastina (factor III), lo que conduce a la liberación proteolítica de la convertina (VIIa).
El complejo FT-VIIa facilita la generación de los factores Xa y IXa y esto causa la producción de una pequeña cantidad de trombina que inicia el proceso. La fase de amplificación se centra en la superficie de las plaquetas, activadas por la trombina, que acumulan en su superficie factores y cofactores y permiten su ensamblaje, dando lugar a complejos con actividad enzimática, como tenasa o protrombinasa, que facilitaran la fase de propagación, con la formación de grandes cantidades de trombina que favorecen la generación de fibrina y su polimerización, para dar lugar al coagulo estable, según se indica en la figura 1.
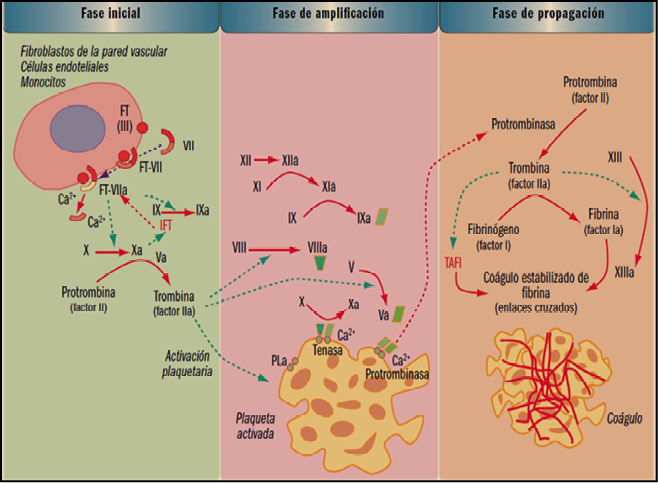
Los trastornos asociados a una deficiencia congénita en la producción de determinados factores de la coagulación sanguínea constituyen uno de los ejemplos típicos de las metabolopatías congénitas de mayor impacto. Aunque se trata de patologías poco frecuentes, su incidencia en varias de las familias reales europeas – debido a la estrecha endogamia – las popularizó.
La más común de las coagulopatías hereditarias – en realidad, la menos infrecuente – es la enfermedad de von Willebrand, un trastorno congénito transmitido autosómicamente, caracterizado por el déficit cualitativo y/o cuantitativo del factor de von Willebrand (Factor vW). Se ha calculado que en determinadas áreas puede llegar a afectar al 1% de la población, aunque los pacientes con problemas hemorrágicos graves no son numerosos. El déficit de factor VIII se conoce como hemofilia A y afecta 1 de cada 10.000 personas a nivel mundial; por tanto, se puede considerar como una enfermedad rara (menos de 5 casos por 10.000 habitantes, en la Unión Europea), al igual que el resto de la coagulopatías hereditarias, aún menos frecuentes que la hemofilia A, salvo la mencionada enfermedad de von Willebrand. Por su parte, la deficiencia de factor IX se conoce como hemofilia B y afecta a uno de cada 30.000 varones. Según la Sociedad Española de Hematología y Hemoterapia (SEHH), en 2016 había en España alrededor de 3.000 pacientes con hemofilia (un 85% con hemofilia A y un 15% con hemofilia B); en el caso de la hemofilia A, el 96% de los casos se trata de varones y un 94% de los casos de hemofilia B.
Todas estas coagulopatías tienen una herencia autosómica recesiva. Clínicamente se expresan con hemorragias de intensidad variable, manifestándose una mayor gravedad en los casos homocigóticos, en los que existe una muy baja concentración de factor de coagulación. Los dos tipos principales de hemofilia (A y B) se transmiten de forma recesiva ligada al sexo (concretamente, en el cromosoma X del par 23), de tal forma que el 100% de las hijas de hemofílicos son portadoras y el 50% de los hijos de mujeres portadoras manifiestan la forma clínica de la hemofilia.
Existe un buen número de enfermedades genéticas ligadas a la pareja cromosómica sexual (XX en las mujeres y XY en los varones). Como los hombres la pareja es heterogénea (XY) sólo tienen una copia de los genes de cada uno de los dos cromosomas (X e Y), por lo que no existen otra copia del gen que puedan contrarrestar la función defectuosa del alelo anómalo situado en dicho cromosoma. Este es el caso de las hemofilias.
La inversión en la secuencia de ADN que se encuentra en el intrón 22 del gen del factor VIII es la anomalía molecular más frecuentemente detectada en pacientes con hemofilia A grave, siendo responsable de la enfermedad en el 45% de los casos. Aproximadamente el 50% de hemofílicos con afección grave y la práctica totalidad de hemofílicos con diátesis hemorrágica moderada o leve presentan mutaciones puntuales. En cambio, en la hemofilia B las deleciones parciales o completas del gen representan el principal defecto molecular. La existencia de mutaciones puntuales es responsable de las formas variantes de enfermedad (alteraciones funcionales). También se han identificado un importante número de alteraciones moleculares responsables de la hemofilia B.
El gen humano del Factor VIII (F8) se encuentra en la banda más distal (Xq28) del brazo largo del cromosoma X y está formado por una secuencia de 186.000 bases (186 kb) dividida en 26 exones y 25 intrones. El Factor VIII se expresa principalmente en el hígado, en las células endoteliales sinusoidales. La proteína precursora del Factor VIII contiene dominios de homología interna y su estructura desde el extremo amino (-NH2) al carboxilo (-COOH) sigue una secuencia A1-A2-B-A3-C1-C2, en la que el dominio B es escindido para formar el Factor VIII, que es activado por la trombina (Cuéllar, 2013).
La hemofilia A puede aparecer como consecuencia de muy diversas mutaciones en el gen F8, de las que aproximadamente un tercio son de novo, es decir, en pacientes sin historial familiar, como ocurre también en la hemofilia B. Entre estas mutaciones se incluyen inversiones (por ejemplo, la inversión de intrón 22); supresiones grandes – que se asocian con formas clínicas graves de hemofilia y un mayor riesgo de anticuerpos inhibidores –, inserciones, duplicaciones y reordenamientos cromosómicos. Las mutaciones puntuales, deleciones o inserciones pequeñas y mutaciones sin sentido están vinculados a formas clínicas específicas, de relevancia algo menor.
La expresión clínica más característica de todas las formas de hemofilia es la hemorragia, que se manifiesta en múltiples niveles y localizaciones orgánicas: muscular, sistema nervioso central, partes blandas y, muy especialmente, en las articulaciones. La gravedad de las manifestaciones clínicas suele estar en relación con la cantidad de factor existente:
- Forma grave. Cuando la actividad funcional del factor de la coagulación es indetectable, es decir inferior al 1%. Habitualmente se trata de pacientes con sangrado espontáneo antes de los 6 meses de edad o hemorragia intracraneal en el parto.
- Forma moderada. Los niveles de factor se encuentran entre el 1 y el 5% de lo normal. El sangrado aparece generalmente antes de los dos años de edad, tras producirse traumas mínimos o pequeñas maniobras exploratorias invasivas.
- Forma leve. Los niveles de factor VIII o IX son superiores al 5% e inferiores al 40%. El sangrado es raro y puede aparecer ante traumatismos importantes o tras intervenciones quirúrgicas.
La hemartrosis o hemorragia articular es la forma de sangrado más frecuente (65-90%), hasta el punto de que constituye el sello distintivo de las hemofilias. En pacientes con hemofilia A o B grave, más del 90% de todos los episodios hemorrágicos se producen en las articulaciones, y 80% de estos representan hemartrosis de los tobillos, rodillas y codos; es menos habitual en la articulación del hombro. En cualquier caso, la hemartrosis produce dolor, tumefacción e impotencia funcional.
Por detrás de la hemartrosis, en orden de frecuencia, los hematomas musculares suponen el 30% de las complicaciones hemorrágicas. Pueden complicarse con síndromes compartimentales e incluso shock hemorrágico; a la larga producen atrofia muscular. Finalmente, la hemorragia intracraneal es la complicación más grave, pero apenas constituye entre el 2 y 13% de las complicaciones hemorrágicas. Si no hay un tratamiento rápido puede causar la muerte. Los hemofílicos también pueden presentar complicaciones hemorrágicas en otras localizaciones, destacando la hematuria y la hemorragia gastrointestinal y orofaríngea.
La incidencia de sangrado en los pacientes con hemofilia durante el período neonatal oscila entre un 20% y un 44%, mientras que la hemorragia intracraneal (HIC) aparece en un 3,5-4% de los neonatos con hemofilia, aunque la cifra podría ser mayor si se incluyen las asintomáticas.
Hace ya más de 150 años que se conocen los beneficios de la transfusión de sangre para controlar la hemorragia relacionada con hemofilia y hace casi un siglo (1923), que Feissly demostrara la superioridad de plasma sobre la sangre total para este fin. La introducción en la década de 1970 de los concentrados con factores derivados del plasma permitió un tratamiento mucho más eficaz y funcional, así como una notable reducción de la morbilidad y mortalidad asociadas a la hemorragia.
Esta favorable evolución del tratamiento de restauración o de reemplazo de los factores sanguíneos implicados en las coagulopatías hereditarias sufrió a principios de la década de los años 80 del pasado siglo un grave revés. Nada menos que tres de cada cuatro pacientes con hemofilia grave acabaron infectados por el virus de la inmunodeficiencia humana (VIH) y prácticamente todos con el de la hepatitis C (VHC), como consecuencia de la transfusión de concentrados de plasma contaminados. En las décadas siguientes, la seguridad de estos productos se convirtió un elemento clave y derivó en la producción de concentrados no contaminados biológicamente, gracias a la incorporación de dobles sistemas de inactivación viral. A ello, cabe agregar la disponibilidad de análogos recombinantes de los factores naturales, carentes por completo de riesgo de contaminación por virus humanos.
Sin embargo, tanto los medicamentos de origen extractivo, a partir de plasma humano, como los de origen recombinante siguen teniendo un problema importante que puede limitar o incluso anular su utilidad: la producción de inhibidores en la sangre de los pacientes.
Los objetivos principales de la terapia son prevenir la hemorragia y en su caso tratarla, así como sus complicaciones y secuelas, restaurar y mantener la función articular e integrar a los pacientes en la vida social normal. Para ello, los hemofílicos A moderados y leves se pueden tratar con desmopresina y antifibrinolíticos en un buen número de ocasiones; por el contrario, en las formas graves, es preciso utilizar un tratamiento sustitutivo con factor VIII o IX, según sea el caso, el cual puede ser concentrado plasmático purificado o recombinante. En todos los casos, la cantidad de factor a infundir depende de la gravedad de la hemorragia. Así, para combatir hemorragias graves o de riesgo vital se debe alcanzar un 100% de factor circulante. Ante una hemartosis, el tratamiento debe ser lo más precoz posible (antes de 4 horas) y ante la duda, siempre se debe tratar. El objetivo de factor a conseguir es de un 30-50% (habitualmente 20-40 U/kg de factor VIII y 30-60 U/kg de factor IX).
En recién nacidos con sospecha de hemofilia se recomienda administrar vitamina K por vía intravenosa a través de los vasos umbilicales. En caso de que no sea posible, otra alternativa es la vía oral, con una dosis de carga de 1 mg, seguida de 25 µg durante varios días.
La desmopresina es un análogo sintético de la vasopresina capaz de liberar del endotelio Factor vW vascular al torrente circulatorio, con aumentos de 4-5 veces su valor basal y durante 8-10 horas. Los efectos adversos son ligeros, habiéndose descrito enrojecimiento facial, cefalea, hipotensión y taquicardia. La desmopresina se administra por vía endovenosa o intranasal y debe considerarse como primera opción terapéutica en pacientes con hemofilia A leve y hemorragias leves o moderadas o procedimientos invasivos menores. Para las hemorragias mucocutáneas se ha mostrado especialmente útil la administración de ácido tranexámico.
La actividad de los factores de coagulación se expresa en unidades internacionales (UI), que corresponden al 100% de actividad del correspondiente factor en 1 ml de plasma de donantes sanos. La administración de 1 UI/Kg de peso aumenta 1-2% de su nivel en plasma. En general, los pacientes con enfermedad grave aumentan 1% después de la primera inyección mientras que el aumento del 2% solo se produce cuando se consigue un equilibrio entre los compartimentos intra y extravascular. En cualquier caso, los regímenes de dosificación habituales se basan en ajustes según el peso de los pacientes, pero la mejor comprensión de la respuesta farmacocinética de un individuo ha demostrado ser más eficaz en la predicción de los niveles de factor de coagulación que protegen contra episodios de sangrado.
El desarrollo de la artropatía hemofílica está directamente relacionado con el número de episodios de sangrado, aunque algunos estudios sugieren que el deterioro de las articulaciones puede ocurrir incluso sin evidencia clínica de hemartrosis. La artropatía, una vez establecida, es irreversible y progresiva; por lo tanto, la profilaxis, definida como la administración a largo plazo regular de los correspondientes factores implicados para prevenir hemorragias articulares, constituye el eje central de la gestión para los niños con hemofilia severa.
La profilaxis primaria consiste en la infusión regular de concentrados del factor deficitario, que se mantiene durante más de 46 semanas al año, y es iniciada antes de la aparición de alteraciones articulares. Hay datos clínicos que demuestran que la profilaxis iniciada a temprana edad protege contra el daño de las articulaciones y disminuye la frecuencia de las hemartrosis y otras hemorragias.
Como ya se ha indicado, el desarrollo de un inhibidor es la complicación más importante del tratamiento de restauración en el paciente hemofílico. Se trata de una inmunoglobulina IgG policlonal - la más común es la de tipo IgG4 – que se une a los dominios funcionales del Factor VIII e impide la interacción con los factores de la coagulación relacionados. Los inhibidores que reconocen epítopos propios del dominio A2 (aminoácidos 484-508) o A3 (aminoácidos 1811-1818) suelen bloquear de forma directa los lugares de unión de alta afinidad del Factor VIII activado con el Factor IX activado y el Factor X, impidiendo la formación del complejo tenasa. Los inhibidores dirigidos contra el dominio C2 (aminoácidos 2181 y 2243) suelen interferir en la unión del Factor VIII a fosfolípidos y al Factor von Willebrand. En general aparece tras las primeras exposiciones al factor – por término medio al cabo de 10-12 días de exposición – y con una frecuencia del 20-30% en los pacientes con hemofilia A grave.
Es recomendable realizar una detección precoz mediante la realización de determinaciones analíticas cada cinco dosis administradas, hasta la vigésima dosis y, posteriormente, cada 10 dosis hasta los 50 días. Superadas las 150 dosis, el riesgo de que aparezca baja notablemente, por lo que es suficiente un control anual. También se realiza una determinación antes de una intervención quirúrgica cuando se cambie el tipo de concentrado administrado, y si se observa ineficacia del tratamiento en el control de un episodio hemorrágico.
La elección del tratamiento hemostático está condicionada por el nivel o título del inhibidor (alto o bajo), y la respuesta después de una nueva estimulación. Se considera como inhibidor de título bajo aquél por debajo de 5 unidades Bethesda por mililitro (UB/ml), que se mantiene a pesar de la estimulación con sucesivas administraciones de Factor VIII. Se considera además como de bajo título y alta respuesta cuando se observa una respuesta rápida (alrededor de siete días) después de la administración de una dosis. Los pacientes con inhibidor de bajo título y poco respondedores son tratados con dosis más elevadas de Factor VIII, y un control estrecho de sus niveles. Para el cálculo de las dosis necesarias se debe tener en cuenta que se precisa una cantidad para neutralizar el efecto del inhibidor, más la cantidad de factor necesario para alcanzar los niveles terapéuticos deseados, de ahí la necesidad de un control estricto y personalizado. En pacientes con título alto puede disminuir el título en ausencia de administración de Factor VIII durante periodos prolongados de tiempo. En caso de pacientes con título alto de inhibidor deben ser tratados con agentes capaces de inducir la hemostasia en ausencia de Factor VIII o Factor IX.
La mayoría de los pacientes con hemofilia A sometidos a un tratamiento profiláctico reciben tres dosis semanales o una dosis cada dos días de 25-40 UI/kg, aunque, es habitual el tratamiento a demanda.
ACCIÓN Y MECANISMO
El efmoroctocog alfa es una proteína de fusión recombinante con una elevada semivida de eliminación, formada por el factor VIII de coagulación humano con deleción del dominio B, unido al dominio Fc de la inmunoglobulina humana G1 (IgG1). Ha sido autorizada para el tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A.
El complejo formado por el factor VIII y el von Willebrand (vW) es escindido al activarse la cascada de coagulación, transformándose el factor VIII en factor VIII activado (VIIIa) y liberando el factor vW; a partir de ese proceso, el factor VIIIa actúa como cofactor del factor IXa, lo cual facilita la activación del factor X a Xa, el cual, a su vez, convierta la protrombina en trombina y ésta el fibrinógeno en fibrina, responsable último de la constitución del coágulo sanguíneo.
ASPECTOS MOLECULARES
El efmoroctocog alfa es una proteína de fusión de origen recombinante formada por la combinación del factor VIII de coagulación en el que se ha suprimido el dominio B, unido covalentemente al dominio Fc de la IgG1. La proteína de fusión tiene un peso molecular aproximado de 220 kDa. La consecuencia de la fusión del factor VIII con un dominio de la IgG1 es un incremento del 50% de la semivida de eliminación frente al factor VIII recombinante (19,0 vs. 12,4 h), lo que permite un efecto más mantenido y una administración menos frecuente que las formas convencionales de factor VIII.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del efmoroctocog han sido adecuadamente contrastadas en la indicación autorizada mediante dos ensayos clínicos de fase 3 (A-LONG y Kids A-LONG), confirmatorios de eficacia y seguridad, y una extensión de ambos aún en desarrollo (ASPIRE).
El estudio A-LONG (Mahlangu, 2014) evalúo la seguridad, eficacia y farmacocinética del efmoroctocog para la profilaxis, el tratamiento de la hemorragia aguda y el control hemostático perioperatorio en 165 varones adultos o adolescentes (≥12 años) con hemofilia A grave, tratados previamente. Se establecieron tres brazos de tratamiento: profilaxis individualizada dosis de 25-65 UI/kg cada 3-5 días (n=118), profilaxis semanal con dosis fijas de 65 UI/kg (n=24) y tratamiento episódico con dosis de 10-50 UI/kg (n=23). Como variable se establecieron la tasa anualizada de sangrado (TAS), el desarrollo de inhibidor del Factor VIII y la incidencia de eventos adversos. Los resultados mostraron que la mediana de la tasa anualizada de sangrado fue de 1,6 (profilaxis individualizada, brazo 1), 3,6 (profilaxis semanal, brazo 2) y 33,6 (tratamiento episódico, brazo 3). En el brazo 1, la mediana de la dosis semanal fue de 77,9 UI/kg y aproximadamente el 30% de los pacientes alcanzó un intervalo de dosificación de 5 días (últimos 3 meses en estudio). No se observó ningún episodio hemorrágico en el 45% de los pacientes sometidos a profilaxis individualizada, ni en el 17% de los tratados con el régimen semanal con dosis fijas. El control de los episodios hemorrágicos se consiguió en el 87% de los casos con una única inyección de efmoroctocg y en el 98% con un máximo de dos.
Por su parte, el estudio Kids A-LONG (Young, 2015) incluyó a 71 pacientes pediátricos (<12 años) con hemofilia A grave, previamente tratados. El régimen posológico inicial de efmoroctocog fue de 25 UI/kg el primer día, 50 UI/kg el 4º, ajustándose la posología definitiva hasta un máximo de 80 UI/kg en intervalos de al menos dos días. La variable primaria fue el desarrollo de inhibidor (anticuerpo neutralizante), mientras que entre las secundarias se determinó la tasa anualizada de sangrado y el número de infusiones requerido para controlar una hemorragia. Los resultados mostraron que ninguno de los pacientes desarrollaron inhibidor, mientras que la tasa anualizada de sangrado fue de 1,96 globalmente pero de 0,0 para hemorragias espontáneas; de hecho, un 47% de los pacientes no experimentó ningún episodio hemorrágico; el 93% de los episodios hemorrágicos fueron controlados con una o dos infusiones de efmoroctocog. La dosis media semanal profiláctica fue de 88,1 UI/kg y al final del estudio, el 90% de los pacientes estaba utilizando una pauta de dos infusiones semanales. Entre aquellos que habían recibido anteriormente un régimen profiláctico con factor VIII, el 74% redujeron la frecuencia de la administración con efmoroctocog.
Finalmente, el estudio ASPIRE (Nolan, 2016) es un estudio de extensión en el que se incluyeron pacientes procedentes de los dos ensayos clínicos anteriores (150 adultos y adolescentes, y 61 niños), que fueron incluidos en uno de los siguientes brazos de tratamiento: profilaxis individualizada, profilaxis semanal, profilaxis modificada (para aquellos pacientes en los que no se consiguió anteriormente una respuesta óptima con la profilaxis individualizada o semanal) y tratamiento episódico. Como se indicó anteriormente, el estudio aún sigue en curso; no obstante, se ha realizado un análisis intermedio de resultados, cuando llevaban transcurridos una mediana de 81 semanas con los adultos y adolescentes, y de 24 semanas con los niños, teniendo la mayoría de los pacientes (92% de adultos y adolescentes, y 57% de los niños) al menos 100 días acumulados de exposición al fármaco. Los resultados en este punto indican que las tasas anualizadas de sangrado en los pacientes sometidos a profilaxis individualizada eran de 0,66 (adultos/adolescentes), 0,0 (niños <6 años) y 1,54 (niños 6-12 años); en los sometidos a profilaxis semanal y modificada fue de 2,0 (adultos/dolescentes). No se observó ninguna variación en la frecuencia o en la dosis de infusión profiláctica en la mayoría de los pacientes, tanto adultos como niños.
Desde el punto de vista de la seguridad, el efmoroctocog presenta un perfil toxicológico francamente benigno. Los eventos adversos más comunes que aparecieron durante el tratamiento fueron nasofaringitis (19%), infecciones del tracto respiratorio superior (12%), artralgia (12%), cefalea (11%), tos (10%), diarrea (5,0%), pirexia (5,6%) y vómitos (5,2%). La mayoría de estos eventos adversos fueron considerados como no relacionados con el tratamiento, salvo los casos documentados de malestar, cefalea, artralgia y mialgia, descritos en un 0,2% de los pacientes. Por su parte, un 16,7% de los pacientes tratados experimentaron eventos adversos graves, aunque ninguno de ellos fueron atribuibles al tratamiento con moroctocog. Solo se ha descrito un único caso de aparición de anticuerpos inhibidores, con baja titulación y que además acabo por negativizarse al cabo de varias semanas.
ASPECTOS INNOVADORES
El efmoroctocog alfa es una proteína de fusión recombinante con una elevada semivida de eliminación, formada por el factor VIII de coagulación humano con deleción del dominio B, unido al dominio Fc de la inmunoglobulina humana G1 (IgG1). Ha sido autorizada para el tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A.
Los datos acumulados de los dos estudios de fase III disponibles y su estudio de extensión, que todavía está en curso, han demostrado la eficacia a largo plazo de efmoroctocog alfa en el tratamiento de episodios agudos de sangrado, el manejo perioperatorio y la profilaxis en varones con hemofilia A grave tratados previamente. Entre los pacientes en los que se utilizó efmoroctocog alfa para la profilaxis individualizadas y que previamente habían recibido profilaxis con factor VIII, prácticamente todos los adultos y adolescentes, y tres cuartas partes de los niños redujeron su frecuencia de infusión en comparación con el régimen utilizado anteriormente. La sustitución del factor VIII por efmoroctocog alfa parece ser bien tolerado en pacientes previamente tratados, sin que se evidencie ningún de aumento de la inmunogenicidad.
Por su parte, la incidencia de eventos adversos asociados al medicamento es mínima, habiéndose documentado algunos cuadros leves de malestar, cefalea, artralgia y mialgia, descritos en un 0,2% de los pacientes. Y, lo que es de especial importancia, solo se ha descrito un único caso de aparición de anticuerpos inhibidores, con baja titulación y que además acabo por negativizarse al cabo de varias semanas.
Aunque no se dispone de estudios directamente comparativos, la evidencia proporcionada por los ensayos clínicos disponibles indica que el efmoroctocog alfa constituye una alternativa eficaz a las preparaciones convencionales de factor VIII, incluyendo las formas recombinantes para el tratamiento de la hemofilia A. Por otra parte, mediante la reducción de la frecuencia de las inyecciones, teóricamente podría mejorarse la adherencia a los regímenes profilácticos (Frampton, 2016). En este sentido, puede considerarse como el primer tratamiento para la hemofilia A capaz de ofrecer una protección prolongada frente a los episodios de sangrado mediante inyecciones profilácticas cada 3 a 5 días.
|
VALORACIÓN |
|
EFMOROCTOCOG ALFA
|
|
Grupo Terapéutico (ATC): B02BD. SANGRE Y ÓRGANOS HEMATOPOYÉTICOS. Vitamina K y otros hemostáticos: factores de coagulación. |
|
Indicaciones autorizadas: Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A. |
|
INNOVACIÓN moderada. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |

|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Factor VIII |
Beriate |
CSL Behring |
2000 |
|
Octocog alfa |
Advate |
Baxalta |
2004 |
|
Moroctocog alfa |
Refacto AF |
Pfizer |
2011 |
|
Simoctocog alfa |
Nuwiq |
Octapharma |
2014 |
|
Efmoroctocog alfa |
Elocta |
Swedish Orphan Viovitrum |
2016 |
BIBLIOGRAFÍA
Bibliografía
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Hemofilia y otras coagulopatías hereditarias. Panorama Actual Med. 2013; 37(362): 232-42.
- European Medicines Agency (EMA). Elonta®. European Public Assessment Report (EPAR). EMA/660642/2015; EMEA/H/C/003964. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003964/WC500198644.pdf
- Frampton JE. Efmoroctocog Alfa: A Review in Haemophilia A. Drugs. 2016; 76(13): 1281-91. doi: 10.1007/s40265-016-0622-z.
- Mahlangu J, Powell JS, Ragni MV, Chowdary P, Josephson NC, Pabinger I, et al; A-LONG Investigators. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014; 123(3): 317-25. doi: 10.1182/blood-2013-10-529974.
- Nolan B, Mahlangu J, Perry D, Young G, Liesner R, Konkle B, et al. Long-term safety and efficacy of recombinant factor VIII Fc fusion protein (rFVIIIFc) in subjects with haemophilia A. Haemophilia. 2016; 22(1): 72-80. doi: 10.1111/hae.12766.
- Young G, Mahlangu J, Kulkarni R, Nolan B, Liesner R, Pasi J, et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. J Thromb Haemost. 2015; 13(6): 967-77. doi: 10.1111/jth.12911.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
