Dexmedetomidina DEXDOR® (Orion)
 Nº377
Nº377

SEDOANALGESIA EN LA UCI
La sedoanalgesia es uno de los elementos fundamentales para el manejo de los pacientes en estado crítico. La mayoría de estos que son ingresados en una Unidad de Cuidados Intensivos (UCI) requiere una combinación de analgesia y sedación prolongadas para reducir su grado de estrés y facilitar su manejo clínico, lo que redundará en un mejor pronóstico.
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (∝2) adrenérgicos, estrechamente relacionado con la clonidina y la oximetazolina, que ha sido autorizado para la sedación de pacientes adultos en la UCI que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond. Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta.
Los ensayos clínicos con dexmedetomidina controlados con midazolam y propofol han mostrado la no inferioridad frente a estos en la indicación autorizada, en particular en lo que se refiere al porcentaje del tiempo (60-65%) durante el cual se mantiene el nivel de sedación requerido para cada paciente, sin necesidad de medicación sedante de rescate; tampoco se han registrado diferencias significativas en cuanto a la duración media del uso de ventilación mecánica en la UCI.
La dexmedetomidina representa una forma algo diferente de sedación para la UCI que los habitualmente utilizados, el propofol y el midazolam. Produce un tipo de sedación consciente, con la que el paciente parece estar dormido pero es fácilmente despertado, manteniendo la cooperación y la comunicación con el personal sanitario. En cualquier caso, supone una nueva vía de actuación con unos perfiles de utilidad y de seguridad propios, lo que en un ámbito sin demasiadas opciones permite ampliar las disponibles para adecuarse a las condiciones específicas de cada paciente.
La sedoanalgesia es uno de los elementos fundamentales para el manejo de los pacientes en estado crítico. La mayoría de estos que son ingresados en una Unidad de Cuidados Intensivos (UCI) requiere una combinación de analgesia y sedación prolongadas para reducir su grado de estrés y facilitar su manejo clínico, lo que redundará en un mejor pronóstico.
El dolor en el paciente crítico es común (hasta el 40% de los pacientes refiere haber sufrido dolor en algún momento de su estancia en la UCI) y contribuye decisivamente a la aparición de trastornos del sueño, agotamiento, desorientación y agitación, y desencadena una respuesta neuroendocrina (respuesta al estrés), con taquicardia, aumento del consumo miocárdico de oxígeno, hipercoagulabilidad, inmunosupresión y aumento del catabolismo proteico, hasta el punto de que la respuesta al estrés es asociada con un aumento de la morbilidad y la mortalidad. Por ello, la sedoanalgesia constituye una parte decisiva de la asistencia integral en la UCI y, de hecho, los fármacos usados con este fin son, junto con los antiulcerosos y los antibacterianos, los más utilizados en el paciente crítico. En definitiva, una adecuada sedoanalgesia reduce la respuesta al estrés, limita la ansiedad, mejora la tolerabilidad a la ventilación mecánica y facilita los cuidados del paciente. Por consenso, se define la sedación prolongada como aquélla cuya duración excede las 72 horas (Estébanez, 2008).
Obviamente, las necesidades de sedación no son las mismas para todos los pacientes, ni para el mismo paciente a lo largo del día ni durante su evolución en la UCI, por lo que se debe individualizar el tratamiento en función de los requerimientos de analgesia y sedación que precise el paciente en cada momento. Normalmente, se consideran don niveles de sedación, el superficial y el profundo.
En la sedación superficial, (con una puntuación 0 a -3 en la Richmond Agitation Sedation Scale, RASS) el objetivo es mantener a los pacientes despiertos o con un grado de sedación que permita un fácil despertar, lo que posibilita una evaluación más fácil del dolor, un mejor contacto del paciente con el personal sanitario y la familia y una cooperación en técnicas como la fisioterapia respiratoria o la evaluación neurológica. Por su parte, la sedación profunda (RASS de -4 a -5) se busca en pacientes en ventilación mecánica en los que es importante inhibir el estímulo respiratorio, aquellos que requieren el uso de bloqueantes neuromusculares, pacientes con hipertensión endocraneal, estatus epiléptico o psicosis aguda; pacientes en los que se realiza limitación del esfuerzo terapéutico y aquellos en los que se realizan técnicas diagnósticas y/o terapéuticas muy agresivas.
Las características del paciente crítico en la UCI requieren idealmente fármacos sedantes con un rápido inicio de acción y una no menos rápida recuperación, un fácil ajuste de la dosificación, un amplio margen terapéutico y la ausencia de acumulación en el organismo, de interacciones farmacológicas y de efectos adversos; todo ello, a un bajo coste. Obviamente, tal medicamento no existe y hay que recurrir a un amplio colectivo de fármacos que cumplen con alguna de las condiciones antes mencionadas, pero no todas, para obtener una sedación prolongada con niveles de eficacia y seguridad razonables. Los utilizados habitualmente son las benzodiacepinas (midazolam, lorazepam, diazepam, etc.), anestésicos (propofol), analgésicos opiáceos (remifentanilo), agonistas α2 adrenégicos (clonidina, etc.) e hipnóticos barbitúricos (tiopental); entre ellos, los más utilizados actualmente son las benzodiazepinas y el propofol.
Las benzodiazepinas presentan unas excelentes propiedades farmacocinéticas para su utilización en el paciente crítico y su empleo es seguro, ya que poseen mayor margen terapéutico que otros hipnóticos y sedantes. Todos los miembros del grupo actúan sobre sobre determinadas subunidades del receptor GABAA que potencian los efectos del ácido γ-aminobutírico (GABA) sobre dicho receptor. En términos bioquímicos, estos fármacos aumentan la respuesta de ciertas neuronas al GABA, facilitando la apertura de los canales de iones cloruro que son activados por este aminoácido neurotransmisor, el cual media gran parte de la transmisión sináptica inhibitoria en el sistema nervioso central. Los fármacos de este grupo dan lugar a un aumento de la frecuencia de apertura del canal inducida por GABA, sin producir cambios en la conductancia o el tiempo medio de apertura. Este efecto es compatible con la observación de que tales fármacos no se fijan al sitio de unión de GABA en el receptor GABAA (sitio ortostérico), sino a un sitio modulador cuya ocupación ocasiona que el GABA incremente su afinidad por el receptor cuando el poro está abierto. Este mecanismo alostérico explica el bajo grado de toxicidad aguda de estos hipnóticos, ya que no pueden ocasionar la entrada de iones cloruro en ausencia de GABA. En términos farmacoterapéuticos, los efectos asociados a la activación de este receptor son ansiolíticos, hipnóticos, amnésicos, miorrelajantes y anticonvulsivantes. Sin embargo, carecen de capacidad analgésica y antiemética. La benzodiazepina más utilizada en sedación prolongada en Europa es el midazolam, pero en principio cualquier miembro del grupo con acción rápida, como el lorazepam, podría utilizarse teóricamente en esta indicación.
Las benzodiazepinas producen disminución de la presión arterial por vasodilatación, además de depresión respiratoria en relación con la dosis, la velocidad de administración y el estado del paciente. Su uso prolongado puede dar lugar al desarrollo de tolerancia. Además, pueden producir conducta agresiva u hostil por desinhibición o un estado inicial paradójico de nerviosismo antes de que se establezca el efecto ansiolítico o sedante. Por el contrario, un aspecto favorable muy interesante de las benzodiazepinas es que ofrecen la posibilidad de revertir su efecto farmacológico mediante la utilización de flumazenilo, un antagonista específico de los receptores, aunque su empleo rutinario en sedación prolongada no está recomendado por el riesgo de aparición de un síndrome de abstinencia.
El propofol o disoprofol tiene propiedades sedantes, hipnóticas y antieméticas, pero carece de efectos analgésicos significativos. Su acción anestésica es consecuencia de su interacción con un lugar alostérico para anestésicos generales en el receptor GABAA, que es diferente al de a las benzodiazepinas, facilitando la apertura del canal de cloruro. Presenta un inicio de acción rápido y una vida media corta. En infusión continua no plantea problemas de acumulación (aunque se ha observado que el tiempo de despertar desde la suspensión del fármaco está relacionado con el tiempo de sedación). Sus características farmacocinéticas permiten un fácil control del nivel de sedación, al igual que una temprana recuperación del nivel de consciencia tras el cese de su administración (unos 10 minutos), lo que facilita la evaluación neurológica de los pacientes. Además, si se asocia a morfina mejora el control de la presión intracraneal en pacientes con traumatismos craneoencefálico graves.
Induce hipotensión por reducción de las resistencias vasculares periféricas sin modificar el gasto cardiaco, lo cual ocurre con más frecuencia tras la administración en bolo, en pacientes hipovolémicos o con inestabilidad hemodinámica. Provoca una profunda depresión respiratoria y bradicardia por depresión del reflejo barorreceptor, en particular, durante la inducción, efecto que es potenciado por los opioides.
Los barbitúricos y, en particular el tiopental (el único utilizado en clínica para esta indicación), actúan de forma similar al propofol (sitio alostérico acoplado a los receptores GABAA del canal de cloruro neuronal), provocando una depresión reversible del tejido nervioso. Producen hipotensión (en bolo) y depresión miocárdica, además de predisponer a la infección y al íleo paralítico, por lo que no se recomiendan para la sedación de rutina de los pacientes críticos, reservándose para aquellos con estatus epiléptico y en pacientes con hipertensión intracraneal refractaria
Entre los analgésicos opiáceos, el más utilizado en sedación prolongada es el remifentanilo. Actúa como un agonista puro de los receptores µ(mu) opiáceos y tanto el inicio de su acción como la duración de ésta son breves. Como es obvio, su acción fundamental es la analgesia, incrementando el umbral del dolor, alterando la percepción del mismo e inhibiendo las vías ascendentes del estímulo doloroso. La sedación con remifentanilo reduce la necesidad de ventilación mecánica y el tiempo de extubación en relación a la sedación estándar y sus propiedades farmacocinéticas lo convierten en un buen agente para su utilización en estrategias de sedación secuencial y dinámica
La utilización de agonistas alfa-2 adrenérgicos no es nueva en sedación, aunque su empleo proviene en origen del ámbito de la veterinaria, donde algunos fármacos de este grupo (xilazina, detomidina) se han venido utilizando durante décadas para inducir la analgesia y la sedación en diversas especies de animales. Todos ellos derivan de la clonidina, aunque la farmacología de este grupo es más compleja de lo que a primera vista pudiera parecer.
Existen tres tipos de receptores o isorreceptores alfa-2 (∝2) adrenérgicos: a, b y c. La acción estimulantes o agonista sobre los receptores ∝2a produce sedación, hipnosis, analgesia e inhibe la secreción de insulina; por su parte, la activación de los receptortes ∝2b produce analgesia espinal y vasoconstricción en la arterias periféricas, mientras que la de los receptores ∝2c da lugar a una modulación del procesamiento sensorial cognitivo y del estado de ánimo, induce una estimulación locomotriz y regula la liberación de noradrenalina por la médula adrenal. Todos estos tipos de receptores poseen localización pre-, post- y extrasinápticas, lo que determina que cada agente activo sobre estos tenga un perfil farmacodinámico particular, no necesariamente extrapolable al resto. Sea como fuere, las localizaciones presinápticas de estos receptores tienen una particular relevancia, por que actúan como autorreceptores, es decir forman parte de un sistema de retroalimentación (feedback) negativo que regula inhibitoriamente la liberación de noradrenalina.
La estimulación a nivel cerebral y espinal de los receptores ∝2 produce una inhibición de la activación (firing) neuronal, que conduce a hipotensión, bradicardia, sedación y analgesia. Reduce la entrada de calcio en las terminaciones nerviosas, lo que puede contribuir a su efecto inhibitorio sobre la liberación de neurotransmisores. En otros órganos y tejidos, produce una reducción de la salivación, y de la secreción y motilidad gástrica; inhibe la liberación de renina, incrementa la tasa de filtración glomerular renal (y con ello la eliminación renal de sodio y agua), reduce la presión intraocular y la secreción de insulina por el páncreas.
Con independencia del fármaco utilizado para producir una sedación prolongada, existe un amplio capítulo de complicaciones asociadas. La primera de ellas es la infrasedación, que desprotege al paciente crítico, haciéndole experimentar miedo, ansiedad, trastornos del sueño, desorientación y agitación, lo que se asocia a un peor pronóstico; todo ello conlleva un incremento del riesgo para la autorretirada (arrancamiento) de dispositivos y de las necesidades de cuidados director de enfermería, lo que se asocia con un aumento del consumo de recursos profesionales y de costes. Además, el aumento del consumo de oxígeno y de la actividad del sistema autónomo con el aumento del trabajo miocárdico puede ser especialmente peligrosos en determinados pacientes críticos (traumatismo craneoencefálico, insuficiencia respiratoria, shock, etc.)
Por el contrario, el empleo de dosis elevadas o de pautas que combinan sedantes puede dar lugar a la sobresedación del paciente, que se asocia a una prolongación del tiempo de ventilación mecánica y, por lo tanto, a las complicaciones relacionadas con la misma (neumonía, hemorragia digestiva, bacteriemia, trombosis venosa profunda, etc.) y a un aumento de la duración de la estancia en la UCI y en el hospital, a un mayor consumo de recursos sanitarios y a la dificultad para monitorizar la evolución neurológica. Asimismo, estos pacientes tienen mayor frecuencia de sueños paranoides, pesadillas y alucinaciones, lo que puede dar lugar a secuelas psicológicas graves.
Por su parte, el desarrollo de tolerancia (requerimiento de dosis progresivamente mayores de sedantes y analgésicos para mantener el mismo nivel de sedoanalgesia) se relaciona con fenómenos de regulación a la baja (down-regulation) de los receptores celulares y da lugar a un aumento de la dificultad para conseguir y/o mantener un nivel adecuado de sedación y, en ocasiones, obliga al empleo de dosis elevadas de sedantes o a la combinación de diferentes fármacos, aumentando el riesgo de complicaciones asociadas
Finalmente, al iniciar la retirada de la sedoanalgesia los pacientes pueden desarrollar síntomas de abstinencia o deprivación1, especialmente si han desarrollado previamente tolerancia o han recibido dosis altas de sedantes durante más de 3-5 días. Los síntomas de abstinencia varían según el fármaco empleado, la edad del paciente, la función cognitiva y la situación clínica. Para ello, en el caso de benzodiazepinas y/u opioides, debe evitarse la suspensión brusca de la pauta, con un descenso progresivo de la dosis de infusión en un 20-40% diario, para continuar con un descenso del 10% cada 12-24 horas guiado por la respuesta clínica, evitando disminuir la dosis en más del 10% en aquellos pacientes con factores de riesgo para el desarrollo de síntomas de abstinencia (especialmente aquellos que han recibido dosis altas durante más de una semana).
ACCIÓN Y MECANISMO
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (∝2) adrenérgicos, autorizado para la sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (Richmind Agitation Sedation Score, RASS).
La dexmedetomidina presenta un amplio abanico de actividades sobre el funcionamiento orgánico: sedación, inducción del sueño, ansiolisis, amnesia, analgesia, hipotensión, bradicardia, etc. Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta. Cuando el fármaco se une y estimula los receptores ∝2 adrenérgicos a este nivel neuronal, se produce una inhibición de la adenil ciclasa, responsable de la formación de AMPc. La reducción de los niveles intraneuronales de AMPc se traduce en un aumento de las actividades anabólicas y catabólicas; simultáneamente, se produce una salida del potasio intracelular a través de los canales de potasio activados por calcio, y una inhibición de la entrada de calcio en las terminaciones nerviosas, todo lo cual produce a una hiperpolarización de la membrana neuronal que suprime la activación del locus ceruleus y la actividad de las vías noradrenérgicas ascendentes. El locus ceruleus es también el origen de las vías adrenérgicas médulo-espinales descendentes, de particular relevancia en los mecanismos reguladores de la transmisión de impulsos nociceptivos (implicados en la sensación de dolor). De hecho, los efectos analgésicos se sitúan fundamentalmente a nivel del asta dorsal de la médula espinal.
La inhibición de la liberación de noradrenalina en el locus ceruleus por la dexmedetomidina impide el control inhibitorio del núcleo preóptico ventrolateral, que libera ácido gamma(γ)aminobutírico (GABA) y galanina, que incrementan la propia inhibición del locus ceruleus y del núcleo tuberomamilar. Esta inhibición también reduce la liberación de histamina y su efecto sobre las neuronas de diversas áreas subcorticales, que se traduce en un efecto hipnótico adicional, tal como ocurre durante el sueño fisiológico.
Los efectos sedantes e hipnóticos de la dexmedetomidina son dependientes de la dosis, hasta el punto de desarrollar una actividad anestésica con dosis elevadas. La amnesia asociada al fármaco es mucho menos marcada que la relacionada con las benzodiazepinas, en particular con midazolam.
La dexmedetomidina suprime los escalofríos (frecuentes durante los periodos postoperatorios), posiblemente por su actividad agonista ∝2b adrenérgica a nivel del centro termorregulador hipotalámico. A pesar de su potente efecto sedante, la dexmedetomidina está asociada como mínimos efectos depresores respiratorios, incluso con dosis muy superiores a las empleadas en sedación.
Los efectos cardiovasculares tienen un carácter bifásico. Tras la administración iv inicial, se produce un aumento de la presión arterial y una bradicardia refleja, como consecuencia de una acción agonista ∝2b adrenérgica en las células musculares lisas vasculares. Este efecto inicial dura entre 5 y 10 minutos, tras los que se produce una ligera caída de la presión arterial como consecuencia de la inhibición del tono simpático central; asimismo, los receptores ∝2b adrenérgicos presinápticos también son activados, lo que da lugar a una reducción de la liberación de noradrenalina, todo lo cual conduce a una reducción de la presión arterial y el ritmo cardiaco.
ASPECTOS MOLECULARES
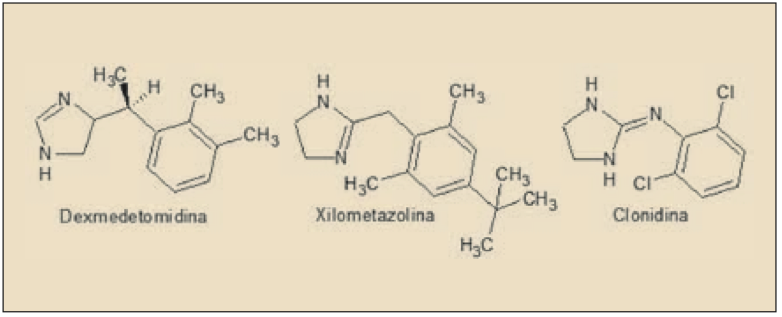
La dexmedetomidina es el enatiómero farmacológicamente activo de la medetomidina. Químicamente, es el 5-[(1S)-1-(2,3-dimetilfenil)etil]-1H-imidazol y está estrechamente relacionado estructural y farmacológicamente con otros agentes agonistas de los receptores α2 adrenérgicos, como clonidina, apraclonidina, brimonidina, nafazolina, tramazolina y, en particular, con xilometazolina y oximetazolina (empleados fundamentalmente con descongestivos nasales). Aunque es considerado fundamentalmente como un agonista de los receptores ∝2 adrenérgicos, dada la presencia de un núcleo de imidazolina en su estructura también desarrolla actividad agonista sobre los receptores imidazolínicos (I), de los que existen tres tipos (I1 implicado en la actividad simpaticolítica de los antihipertensivos imidazolínicos, como la clonidina; I2, una zona de unión alostérica de la monoamino oxidasa, implicada en la modulación del dolor; I3, regula la secreción de insulina por las células beta pancreáticas).
EFICACIA Y SEGURIDAD CLÍNICAS
Existe un buen número de ensayos clínicos controlados con placebo y con comparadores activos que han permitido contrastar la eficacia y la seguridad clínicas de la dexmedetomidina en la indicación autorizada. En el informe público de evaluación de la Agencia Europea de Medicamentos (EMA, 2011) se recogen tres ensayos clínicos principales (pivotales) controlados con comparador activo (propofol y midazolam), todos ellos de fase 3, multicéntricos, multinacionales, aleatorizados y doblemente ciegos, realizados en pacientes sometidos a sedación continua durante más de 24 h (máximo de 14 días) en una Unidad de Cuidados Intensivos (UCI) y con ventilación mecánica.
La dexmedetomidina fue administrada en infusión IV (sin bolo inicial) con un ritmo de 0,8 µg/kg/h la primera hora, reajustándose en función de las necesidades entre 0,25 y un máximo de 1,4 µg/kg/h para mantener el nivel de sedación requerido (expresado como puntuación RASS) para cada paciente. Las dosis de propofol utilizadas variaron entre 0,3 y 4,0 mg/kg/h y las de midazolam entre 0,03 y 0,20 mg/kg/h.
Como criterio de valoración de la eficacia se definieron dos variables co-primarias. La primera fue la proporción de tiempo durante el cual se mantuvo el nivel de sedación requerido para cada paciente sin necesidad de recurrir a medicación sedante adicional (de rescate), mientras que la segunda variable consistió en la duración media de la estancia en la UCI (estudio 3005011) o en la duración media de la ventilación mecánica (estudios 3005012 y 3005013).
En el primero de estos estudios (3005011; n= 79), la mediana del porcentaje del tiempo durante el que se mantuvo la sedación requerida fue del 55,4% (IC95% 48,9 a 65,0) con dexmedetomidina vs. 57,2% (IC95% 47,4 a 66,9) con propofol o midazolam. En aquellos cuyo objetivo era mantener una puntuación RASS entre 0 y -3, estos porcentajes fueron del 67,6 vs. 63,7%, mientras que en aquellos cuyo objetivo era una puntuación de -4 la dexmedetomidina fue claramente inferior a los comparadores (30,7 vs. 63,0%; p= 0,0006). En cuanto a la duración media de la estancia en la UCI fue de 175 vs 187 h (los valores de las medianas fueron de 137 y 132 horas, respectivamente).
En el segundo estudio (3005012; n= 498) se comparó a la dexmedetomidina con propofol, obteniéndose unos valores para el porcentaje de tiempo durante el que se mantuvo el nivel de sedación requerido del 62,4 vs. 65,5% (diferencia ajustada de -2,22; IC95% -7,05 a 2,60), mientras que la mediana de la duración de la ventilación mecánica fue de 96,5 vs. 117,5 h. Un 73% vs. 64% de los pacientes requirieron al menos una vez sedación adicional de rescate (midazolam).
En el tercer estudio (3005013; n= 500) la comparación se hizo con midazolam, registrándose unos valores para el porcentaje de tiempo con el nivel de sedación requerido del 59,9 vs. 54,9% (diferencia ajustada de 4,97; IC95% -0,46 a 10,40), siendo las medianas de la duración de la ventilación mecánica de 123,0 vs. 164,0 h. Un 44% vs. 45% de los pacientes requirieron al menos una vez sedación adicional de rescate (propofol).
En cuanto al perfil de seguridad de la dexmedetomidina, los eventos adversos más comunes descritos son hipotensión, bradicardia e hipertensión (inicial). El porcentaje de pacientes con eventos adversos relacionados con el tratamiento fue (en los ensayos clínicos controlados) del 36% (dexmedetomidina), 25% (midazolam), 20% (propofol) y 27% (placebo), siendo de carácter grave en el 3,0%, 1,0%, 1,7% y 1,5%, respectivamente. Se suspendió el tratamiento por la incidencia de eventos adversos en el 8,2%, 8,7%, 8,4% y 3,6%, respectivamente. Los eventos adversos más prevalentes descritos fueron bradicardia, con 24% (12% con midazolam y 10% con propofol), hipotensión (22%, 17% y 14%), taquicardia (15%, 17% y 1,1%) e hipertensión arterial (12%, 15% y 0%).
ASPECTOS INNOVADORES
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (∝2) adrenérgicos, estrechamente relacionado con la clonidina y la oximetazolina, que ha sido autorizado para la sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (Richmind Agitation Sedation Score, RASS). Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta y que da lugar a una inhibición de la liberación de noradrenalina en dicho núcleo, lo que impide el control inhibitorio del núcleo preóptico ventrolateral, que libera ácido gamma(γ)aminobutírico (GABA) y galanina, que incrementan la propia inhibición del locus ceruleus y del núcleo tuberomamilar. Esta inhibición también reduce la liberación de histamina y su efecto sobre las neuronas de diversas áreas subcorticales, que se traduce en un efecto hipnótico adicional, tal como ocurre durante el sueño fisiológico.
Los ensayos clínicos controlados con midazolam y propofol han mostrado la no inferioridad frente a estos en la indicación autorizada, en particular en lo que se refiere al porcentaje del tiempo (60-65%) durante el cual se mantiene el nivel de sedación requerido para cada paciente, sin necesidad de medicación sedante de rescate; tampoco se han registrado diferencias significativas en cuanto a la duración media del uso de ventilación mecánica en la UCI. Sin embargo, sí se apreció un uso más frecuente de la medicación sedante de rescate con dexmedetomidina que con propofol (73 vs. 64%), pero similar al midazolam (60 vs. 57%). No obstante, cuando se utiliza en pacientes en los que se requiere un nivel de sedación más profundo (puntuación -4 RASS), la desxmedetomidina fue claramente inferior a propofol y midazolam.
Desde el punto de vista de la seguridad, el nuevo fármaco presenta un perfil coherente con so condición farmacológica de agonista ∝2 adrenérgico, siendo los eventos adversos más comunes la hipotensión (con hipertensión inicial) y la bradicardia; en cualquier caso, se trata de fenómenos conocidos y manejables clínica y farmacológicamente.
Un meta-análisis (Fraser, 2013) que incluyó los resultados de seis ensayos clínicos sugiere que el uso de dexmedetomidina y propofol reducen significativamente la duración de la estancia en la UCI, en relación a las benzodiazepinas (midazolam, lorazepam, etc.), así como la necesidad de la ventilación mecánica de los pacientes (diferencia de 1,9 días, IC95% 1,7 a 2,1; p< 0,0001), pero sin diferencias en cuanto a la prevalencia de delirio y a la mortalidad.
Otro meta-análisis (Xia, 2013), confeccionado a partir de los datos de 10 ensayos clínicos controlados, mostró que la dexmedetomidina parece ofrecer ventajas sobre el propofol en términos de la duración de la estancia en la UCI (-0,81 días; IC95% -1,48 a -0,15) y un 60% de reducción en el riesgo de delirio (RR= 0,40; IC95% 0,22 a 0,74), pero no encontró diferencias estadísticamente significativas en cuanto a la duración de la ventilación mecánica (0,53 horas; IC95% -2,66 a 3,72) o mortalidad (RR= 0,83; IC95% 0,32 a 2,12). La dexmedetomidina fue asociada con un mayor riesgo de hipertensión (RR= 1,56; IC95% 1,11 a 2,20).
Se trata de una nueva línea de actuación farmacológica en sedación humana, ya que en veterinaria este tipo de agentes se venía utilizando desde hace décadas; de hecho, la dexmedetomidina es un análogo de la etomidina, profusamente utilizada en veterinaria. En un sentido estricto, no se trata de un medicamento reciente, toda vez que ya se intentó su autorización por la Agencia Europea de Medicamentos (EMA) en 1998, pero ante las objeciones mayores presentadas por ésta, el expediente de registro fue retirado. Posteriormente, con una profunda revisión de dicho expediente e incluyendo nuevos y más rigurosos ensayos clínicos, fue presentado de nuevo en 2010, siendo autorizado el medicamento en 2011 para toda la Unión Europea.
La dexmedetomidina representa una forma algo diferente de sedación para la UCI que los habitualmente utilizados, el propofol y el midazolam (en Estados Unidos se utiliza más habitualmente el lorazepam entre las benzodiazepinas para esta indicación). Produce un tipo de sedación consciente, con la que el paciente parece estar dormido pero es fácilmente despertado, manteniendo la cooperación y la comunicación con el personal sanitario. En cualquier caso, supone una nueva vía de actuación con unos perfiles de utilidad y de seguridad propios, lo que en un ámbito sin demasiadas opciones permite ampliar las disponibles para adecuarse a las condiciones específicas de cada paciente.
|
VALORACIÓN |
|
|---|---|
|
Dexmedetomidina ► DEXDOR® (Orion) |
|
|
Grupo Terapéutico (ATC): N05CM. SISTEMA NERVIOSO. Psicolépticos. Hipnóticos y sedantes: otros. |
|
|
Indicaciones autorizadas: Sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal (correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (RASS)). |
|
|
Valoración global: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad molecular: Mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma o similar indicación terapéutica. |
⇑ |
Bibliografía
Bibliografía
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Estébanez Montiel MB, Alonso Fernández MA, Sandiumenge A, Jiménez Martín MJ; Grupo de Trabajo de Analgesia y Sedación de la SEMICYUC. Sedación prolongada en Unidades de Cuidados Intensivos. Med Intensiva. 2008; 32 Supl 1: 19-30.
- European Medicines Agency (EMA). Dexdor®. European Public Assessment Report (EPAR). EMA/648646/2011; EMEA/H/C/002268. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002268/WC500115632.pdf
- Fraser GL, Devlin JW, Worby CP, Alhazzani W, Barr J, Dasta JF, Kress JP, Davidson JE, Spencer FA. Benzodiazepine versus nonbenzodiazepine-based sedation for mechanically ventilated, critically ill adults: a systematic review and meta-analysis of randomized trials. Crit Care Med. 2013; 41(9 Suppl 1): S30-8. doi: 10.1097/CCM.0b013e3182a16898.
- Xia ZQ, Chen SQ, Yao X, Xie CB, Wen SH, Liu KX. Clinical benefits of dexmedetomidine versus propofol in adult intensive care unit patients: a meta-analysis of randomized clinical trials. J Surg Res. 2013; 185(2): 833-43. doi: 10.1016/j.jss.2013.06.062.
1 El término deprivación es una mala traducción de la voz inglesa deprivation, que aparece en textos científicos. El Diccionario Panhispánico de Dudas recomienda su sustitución por los términos privación, abstinencia, falta o carencia, según los casos.
Artículos relacionados
-
5 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
29 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
6 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
