Carfilzomib (▼Kyprolis®, Amgen) en mieloma múltiple
 Nº399
Nº399

Resumen
El carfilzomib es un antineoplásico que actúa como un inhibidor selectivo e irreversible de la actividad quimotripsina del complejo enzimático proteasoma 20S, lo que induce la acumulación de proteínas poliubiquitinadas proaptóticas y esto, en última instancia, provoca la detención del ciclo celular, facilita la apoptosis e inhibe la tumorigénesis. Ha sido autorizado para el tratamiento, en combinación con lenalidomida y dexametasona o dexametasona sola, de pacientes adultos con mieloma múltiple que han recibido como mínimo un tratamiento previo. La combinación con lenalidomida y dexametasona prolonga en 8,7 meses la supervivencia libre de progresión conseguida con estos dos últimos fármacos, incrementando la supervivencia global a los 24 meses en 8,3 puntos porcentuales (73,3 vs. 65,0%), todo ello con tasas de respuesta global del 87,1 vs. 66,7%, que fueron completas en el 31,8 vs. 9,3%. El carfilzomib es un medicamento que amplía la ya de por sí importante toxicidad de la combinación lenalidomida/dexametasona, aunque no parece que aumente el porcentaje de pacientes que se van obligados a suspender el tratamiento por este motivo; en general, los efectos secundarios más frecuentes son anemia, cansancio, diarrea, trombocitopenia, náuseas, fiebre, disnea, infecciones respiratorias, tos y edema periférico. El carfilzomib supone una evolución en el ámbito de los inhibidores del proteasoma, una herramienta celular fundamental en el control del ciclo vital celular y de la apoptosis, particularmente en el ámbito tumoral. Su efecto irreversible y su mayor selectividad sobe la función enzimática de tipo quimotripsina del proteasoma en relación a su antecesor, el bortezomib, se traduce en un efecto antitumoral más potente y eficaz que el de éste, pero también algo más tóxico. La incorporación del carfilzomib supone un avance moderado pero real en el tratamiento del mieloma múltiple.
ASPECTOS FISIOPATOLÓGICOS
El mieloma múltiple o mieloma de células plasmáticas es un agresivo tumor hematológico maligno de linfocitos B, que presenta algunas características similares a la leucemia. Supone aproximadamente el 1% de los casos de cáncer y, específicamente, el 10% de los hematológicos; sin embargo, dado su actual carácter de incurable, supone el 2% de la mortalidad por cáncer y el 20% de la de los cánceres hematológicos. La incidencia en la Unión Europea es de 4,5-6,0 casos por cada 100.000 habitantes/año y la mortalidad es de 4,1/100.000/año; la supervivencia a los cinco años está en torno al 47%.
La mediana de edad de diagnóstico se sitúa en torno a los 65 años y solo un 2% de los casos diagnosticados corresponden a pacientes menores de 40 años. Es algo más común en varones que en mujeres (1,4:1). En general, los países del sur de Europa – España entre ellos – tienen tasas de incidencia y mortalidad más bajas que los del norte; sin embargo, tanto en unos como en otros la mortalidad por esta patología está creciendo. En España se registran entre 1.500 y 2.000 casos al año (40 nuevos casos por cada millón de habitantes), cifras que alcanzan globalmente en la Unión Europea cerca de 30.000 casos. Su incidencia en España es de 1,5-2,5 por cada 100.000 en menores de 65 años, pero sube a 25-30 a partir de esa edad, afectando a un número ligeramente superior de hombres que de mujeres.
El mieloma múltiple forma parte de las denominadas gammapatías monoclonales, un grupo heterogéneo de enfermedades caracterizadas por una producción anormal de inmunoglobulinas y la aparición de tumores de células plasmáticas. Las células plasmáticas malignas características del mieloma múltiple – células mielomatosas – se acumulan en la médula ósea y pueden llegar al torrente sanguíneo, provocando una alteración de la función de la médula ósea normal, daños óseos y alteración de la función inmune. Las células mielomatosas pueden también formar tumores localizados, los plasmocitomas, que pueden tener una localización tanto ósea como extraósea; justamente es cuando coexiten varios plasmocitomas, con diversas localizaciones, es cuando se emplea apropiadamente el término mieloma múltiple.
La característica típica de la célula mielomatosa es la producción y liberación al torrente circulatorio de una inmunoglobulina monoclonal, denominada proteína M, pero también conocida como proteína mielomatosa, para-proteína o proteína en pico (esto último es debido a la determinación mediante electroforesis). Se trata de una inmunoglobulina producida como consecuencia de la aparición de una o más mutaciones en los genes responsables de la producción de inmunoglobulinas en la célula mielomatosa.
La proteína M tiene una secuencia de aminoácidos y una estructura anormales en relación a las inmunoglobulinas fisiológicas, que provocan la adherencia entre las moléculas y con las estructuras celulares y tisulares tejidos: células sanguíneas, pared de los vasos sanguíneos u otros componentes de la sangre. Todo ello provoca la disminución del flujo sanguíneo, causando un síndrome de hiperviscosidad. En un 30 % de los casos se produce una mayor cantidad de cadenas livianas que las requeridas para combinar con las cadenas pesadas de las inmunoglobulinas, dando lugar a la proteína de Bence-Jones. Esta tiene un peso molecular de 22,000 daltons (Da) y es lo suficientemente pequeña como para ser excretada con la orina y producir un aumento de nivel de proteínas urinarias. Estas proteínas también pueden adherirse entre sí o con otros tejidos (como la inmunoglobulina completa), dando lugar a amiloidosis (depósitos proteicos en cualquier tejido del organismo, como riñón, tejido nervioso o músculo cardíaco) o a la enfermedad de depósito de cadenas livianas (las cadenas livianas se depositan al azar, pero especialmente en los pequeños vasos del ojo o del riñón). Las proteínas monoclonales anormales también tienen pueden provocar un amplio abanico de perturbaciones fisiológicas, al unirse a los factores de la coagulación (dando lugar a trastornos diversos de coagulabilidad) y a otras sustancias circulantes, con diversas consecuencias hormonales y metabólicas (Cuéllar, 2014).
La Beta 2 microglobulina (β2M) es una pequeña proteína cuyos niveles se encuentran elevados en pacientes con mieloma en actividad, aunque un 10% de los pacientes con mieloma no la producen. Precisamente, el estadiaje internacional del mieloma múltiple se basa en las concentraciones en suero de albúmina y de microglobulina β2: el estadio I se caracteriza por niveles de β2M inferiores a 3,5 mg/L y de albúmina mayores de 3,5 g/L; el II por β2M < 3,5 mg/L y de albúmina < 3,5 g/L, o bien β2M de 3,5 a 5,5 mg/L; finalmente, el estadio III se caracteriza por β2M > 5,5 mg/L (Novelli, 2011). Estos estadios se asocian con valores medianos de supervivencia progresivamente inferiores: I (62 meses), II (44) y III (29).
La etiología del mieloma múltiple es poco conocida. No obstante, se ha asociado el mieloma múltiple con la exposición a radiaciones ionizantes. Igualmente, el riesgo parece ser mayor si existen exposiciones ocupacionales relacionadas con la agricultura, refinerías, industrias del corcho, del metal, del plástico o de la madera, o si se ha trabajado como conductor de camiones. Algunos estudios apuntan también hacia la asociación con algunas substancias químicas como asbesto, benceno, pesticidas o pinturas y disolventes. Se ha sugerido la posible relación con algunas enfermedades autoinmunes, como la artritis reumatoide.
El crecimiento descontrolado de las células mielomatosas tiene como una importante consecuencia la destrucción del esqueleto, la insuficiencia de la médula ósea, hipervolemia e hiperviscosidad sanguíneas, supresión de la producción de inmunoglobulinas normales e insuficiencia renal. Sin embargo la enfermedad puede permanecer asintomática durante muchos años. En la fase sintomática, el dolor óseo es el cuadro de presentación más común. Las células mielomatosas y el aumento del número de osteoclastos parecen ser las responsables de la destrucción ósea. Los factores endocrinos locales que regulan la formación y remodelación ósea son una auténtica miríada y consisten mayoritariamente en citocinas y factores de crecimiento producidos por células de los sistemas inmune y hematopoyético. El mecanismo que produce la activación de osteoclastos es complejo, pero se ha observado que participan diversas citocinas locales, tales como interleucinas (IL-1b, IL-6) y factor de necrosis tumoral alfa (TNFα), quimiocinas como las MIP-1 e las integrinas implicadas en el proceso de adhesión celular.
Entre las citocinas implicadas en el mieloma múltiple, quizá la mejor caracterizada sea la IL-6, producida fundamentalmente por las células madre de la médula ósea y los macrófagos. La IL-6 parece ser un importante mediador para el crecimiento, supervivencia y migración celular del mieloma, e incluso de la resistencia a la quimioterapia. Actúa como un estímulo paracrino de las células plasmáticas; por otro lado, la adhesión de las células plasmáticas al estroma de la médula ósea incrementa la secreción por éste de IL-6, lo que forma un auténtico sistema de retroalimentación – un círculo vicioso – que potencia la tumorigénesis plasmática; en definitiva, la IL-6 parece tener un papel fundamental en la patología del mieloma múltiple, potenciando la supervivencia de las células plasmáticas e inhibiendo su apoptosis. El efecto acumulativo de este complejo ambiente bioquímico da lugar a un desequilibrio de las fuerzas anti- y proapoptóticas dentro de las células de mieloma, favoreciendo la expansión clonal desregulada y la proliferación tumoral (Naymagon, 2016).
Actualmente, se considera que los factores efectores finales que regulan la remodelación ósea forman parte de la superfamilia del Factor de Necrosis Tumoral (TNF) y de la de su receptor. Entre ellos puede citarse el RANKL (Ligando del Receptor del Activador del Factor Nuclear Kappa-B), cuya producción es máxima en las células indiferenciadas del estroma osteoclástico y se reduce a medida que madura el fenotipo osteoblástico. Estimula la diferenciación, supervivencia y fusión de las células precursoras de osteoclastos, activa los osteoclastos maduros y prolonga su vida útil. Como resultado permite la expansión de la masa osteoclástica activa capaz de formar sitios de resorción ósea.
El tejido nervioso es afectado con frecuencia en los pacientes con mieloma, tanto por los efectos directos de los anticuerpos de las proteínas mielomatosas frente a la mielina, o por el depósito de fibrillas proteicas (amiloide). Estos efectos resultan en neuropatías periféricas. Por la susceptibilidad a las infecciones, las infecciones virales de los tejidos nerviosos son muy frecuentes, en especial por varicela zoster y parálisis de Bell. Tanto en el hueso como en los tejidos blandos puede producirse una compresión o desplazamiento de los nervios procedentes de la médula espinal o del tallo cerebral.
La predisposición a las infecciones es quizá el rasgo más característico de los pacientes con mieloma junto con la enfermedad ósea, debido a la inhibición de las funciones inmunes normales: producción deficitaria de anticuerpos normales, daño de la función de los linfocitos T y activación anómala de la función monocito/macrófago. Los pacientes con mieloma son particularmente susceptibles a las infecciones virales y a las infecciones con bacterias encapsuladas como el neumococo.
El tratamiento se basa fundamentalmente en la supresión de las células mielomatosas mediante quimio y/o radioterapia, asociado eventualmente a trasplante de células progenitoras hematopoyéticas periféricas. El fármaco antineoplásico más comúnmente utilizado es el melfalán, que continúa siendo el mejor agente único para el tratamiento del mieloma múltiple. La mayoría de los pacientes responden, particularmente cuando se lo utiliza combinado con prednisona. La asociación melfalán/prednisona (protocolo MP) es utilizada frecuentemente, dando lugar a respuestas objetivas en el 60% de los pacientes, manifestada por un 50% mejora en los niveles de proteína M, en los recuentos sanguíneos y en otros resultados bioquímicos, además de la mejoría de varios síntomas de la enfermedad, como el dolor óseo y la fatiga. La ciclofosfamida puede reemplazar el melfalán ya que tiene una actividad antimielomatosa similar, aunque está menos experimentada. Asimismo, en pacientes con mieloma múltiple, los resultados alcanzados con bendamustina son, como mínimo, equiparables a los obtenidos con melfalán; incluso, la tasa de respuestas completas es sustancialmente mayor con bendamustina y el porcentaje de pacientes supervivientes a los cinco años también (29% vs. 19%).
Aunque se han desarrollado combinaciones quimioterápicas más complejas con respuestas similares, o incluso ligeramente mejores, no está claro que sean de primera elección. De hecho, el protocolo MP sigue considerándose como el estándar terapéutico de inicio, dejando las combinaciones más complejas como una segunda línea para aquellos pacientes que no alcanzan una respuesta satisfactoria.
Diferentes líneas de investigación han facilitado la incorporación de otros agentes antineoplásicos, de carácter más selectivo, como es el caso del bortezomib, un inhibidor del proteasoma 26S, autorizado en 2004 para el tratamiento de los pacientes con mieloma múltiple que han recibido previamente al menos dos tratamientos y que presentan progresión de la enfermedad demostrada con el último de estos tratamientos. Los datos clínicos disponibles indican unas tasas de respuesta del orden del 35% para respuestas completas y parciales, con tiempos de progresión de la enfermedad de 9 a 13 meses para estos pacientes respondedores, bastante superiores a los descritos en la bibliografía para los pacientes con mieloma múltiple refractario o recidivante (alrededor de tres meses).
Algunos estudios premiliminares mostraron que la talidomida es capaz de producir una significativa respuesta en pacientes con mieloma, con tasas de respuesta del orden del 25% en pacientes con mieloma recidivante/refractario, en su mayoría, después de un doble trasplante. El problema que plantea este tratamiento es el de la toxicidad. De hecho, la talidomida comenzó a ser investigada tan solo cuatro años después de su retirada por su desgraciada implicación en numerosos y graves casos de dismorfogénesis fetal, calculándose que entre 1956 y 1962 más de 10.000 niños nacieron con importantes deformidades, fundamentalmente focomelia. Sin embargo, sus interesantes propiedades inmunomoduladoras atrajeron el interés de los científicos y algunos años después se había comprobado su potencial utilidad en múltiples patologías de tipo dermatológico, infeccioso y autoinmune. Asimismo, se comprobó su potencial antiangiogénico, siendo ensayada en diversos modelos de cáncer.
A pesar de haber sido autorizada como medicamento huérfano, incluso para el mieloma múltiple – en 2006 – su toxicidad siempre ha sido un factor limitante de todo su potencial farmacológico. De ahí, la necesidad de fármacos que mantuvieran sus propiedades o incluso las ampliaran, pero limitando su notable perfil toxicológico. Por ello, se desarrollaron derivados como la lenalidomida, que no solo mantenían las propiedades de la talidomida sobre diversos biomarcadores implicados en diversas patologías, sino que incluso las ampliaban, mejorando los resultados clínicos en mieloma múltiple, con un perfil toxicológico algo más benigno.
La pomalidomida es un análogo de talidomida y lenalidomida (de hecho, se trata del oxoderivado de la lenalidomida), utilizado en pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento. Presenta una actividad antimielomatosa mayor que sus antecesores, con un perfil toxicológico similar o incluso algo más favorable.
Un aspecto confuso del tratamiento del mieloma ha sido el descubrimiento de que una disminución de los niveles de la proteína mielomatosa en el suero y/o en la orina no se traslada necesariamente a una remisión o a un aumento de la supervivencia. Dado que ningún tratamiento actual erradica todas las células mielomatosas, las características de aquellas residuales tras la quimioterapia inicial son de particular importancia. En este sentido, unas pocas células agresivas residuales pueden causar más problemas que una gran cantidad de células inactivas.
El empleo de altas dosis de quimioterapia (AD) asociado con el trasplante autólogo de células progenitoras hematopoyéticas (TASPE) ha demostrado mejorar tanto las tasas de respuesta como las expectativas de vida en los pacientes con mieloma. Se han alcanzado tasas de remisión completa que oscilan entre un 25% y un 75%. No obstante, el 90% de los pacientes recaen, con tiempos medios de recaída de 18 a 24 meses. La expectativa global de vida en estos pacientes es de aproximadamente 4 a 5 años.
La radioterapia es una importante de tratamiento del mieloma. Para los pacientes con importante destrucción ósea, intenso dolor y/o compresión nerviosa o de la médula espinal, la radioterapia local puede ser muy efectiva. La mayor desventaja es que daña en forma permanente las células progenitoras hematopoyéticas normales de la médula ósea en el área tratada.
Al margen del tratamiento específico, los pacientes con mieloma requieren una serie de tratamientos de soporte, destinados a paliar o a suprimir las principales complicaciones orgánicas del mieloma. En este sentido, la epoetina o sus análogos son administraodos para mejorar los niveles de hemoglobina en los pacientes que tienen anemia persistente. Los bisfosfonatos son empleados como coadyuvantes en los pacientes con mieloma que tienen problemas óseos. Igualmente, el uso de altas dosis de gammaglobulina puede ser requerido en pacientes con infecciones agudas y severas recurrentes. Por otro lado, los factores de crecimiento de colonias, como el GM-CSF, pueden ayudar a mejorar el recuento de glóbulos blancos en un esfuerzo por evitar complicaciones infecciosas.
Las recaídas que ocurren tras 1-3 años de haberse producido una primera remisión, son un problema frecuente en el mieloma. Aunque el inteferón alfa o la prednisona en el tratamiento de mantenimiento pueden ser útiles para prolongar el período de remisión inicial, la recaída que sobreviene inevitablemente, requiere una reinducción con quimioterapia. Si ocurre una recaída después de una remisión de por lo menos 6 meses a un año, la primera estrategia es reutilizar la terapéutica que ha producido la remisión la primera vez. Aproximadamente el 50% de los pacientes alcanzarán la remisión. Esto ocurre especialmente en los pacientes en remisión de más de un año, tras la inducción inicial. Si la remisión ha durado menos de 6 meses, se deben utilizar algunas terapias alternativas. Éste también es el caso para la recaída que ha ocurrido después de la segunda o tercera vez que se utilizó el esquema inicial.
ACCIÓN Y MECANISMO
El carfilzomib es un antineoplásico que actúa como un inhibidor selectivo e irreversible de la actividad quimotripsina del complejo enzimático proteasoma 20S, lo que induce la acumulación de proteínas poliubiquitinadas proaptóticas y esto, en última instancia, provoca la detención del ciclo celular, facilita la apoptosis e inhibe la tumorigénesis. Ha sido autorizado para el tratamiento, en combinación con lenalidomida y dexametasona o dexametasona sola, de pacientes adultos con mieloma múltiple que han recibido como mínimo un tratamiento previo.
El proteasoma 26S es un complejo multiproteico de gran tamaño presente en el citoplasma y en el interior del núcleo de todas las células eucarióticas. Está formado por dos subunidades, un núcleo central catalítico (20S) y una subunidad reguladora. Está implicado en la degradación celular de las proteínas poliubiquitinadas, la cual es esencial para el mantenimiento de la homeostasis celular: más del 80% de todas las proteínas celulares son procesadas por este complejo enzimático, que está implicado en múltiples funciones de control de la degradación proteica, especialmente de las proteínas reguladores que controlan el ciclo celular y la apoptosis, entre las que cabe destacar, las ciclinas p53 y p27, el Inhibidor Proteico Kappa B (IκB) y el Factor Nuclear Kappa B (NFκB).
Las proteínas destinadas para la degradación son marcadas con cadenas de ubiquitina (una proteína de 76 aminoácidos), que se unen al receptor 19S del proteasoma. Este marcaje de sustratos destinados a la degradación es un proceso regulado por un sistema multienzimático. Una vez identificada la proteína marcada con ubiquitina, esta última es eliminada y la proteína es desnaturalizada mediante un proceso complejo de acciones proteolíticas, que se producen en el núcleo 20S del proteasoma. En éste, existe un canal con tres zonas de actividad proteolítica: tipo tripsina, tipo caspasa (post-glutamil hidrólisis) y tipo quimotripsina. Las funciones proteolíticas del proteasoma se asemejan a las serina proteasas, aunque con la salvedad de que actúan sobre un resto de treonina en la zona activa. Las proteínas procesadas por el proteasoma son fraccionadas en restos peptídicos de entre 3 y 22 aminoácidos. En este sentido, el carfilzomib es entre 50 y 300 veces más selectivo frente a la actividad quimitripsina que sobre el resto de actividades proteolíticas (tipo caspasa o tripsina) del proteasoma.
El efecto inhibidor de la actividad del proteasoma afecta a las células neoplásicas a través de varios mecanismos, pero fundamentalmente como consecuencia de la alteración de las proteínas reguladoras que controlan el ciclo celular y la activación nuclear del Factor Nuclear kappa B (NFκB). La inhibición del proteasoma por el carfilzomib conduce a una detención del ciclo celular y de la apoptosis (autodestrucción celular programada). El NFκB es un factor de transcripción nuclear cuya activación es imprescindible en diversos procesos ligados a la tumorigénesis, entre los que cabe destacar el crecimiento y la supervivencia celulares, la angiogénesis (formación de vasos tumorales), las interacciones entre células de diferentes tejidos y la metástasis.
ASPECTOS MOLECULARES
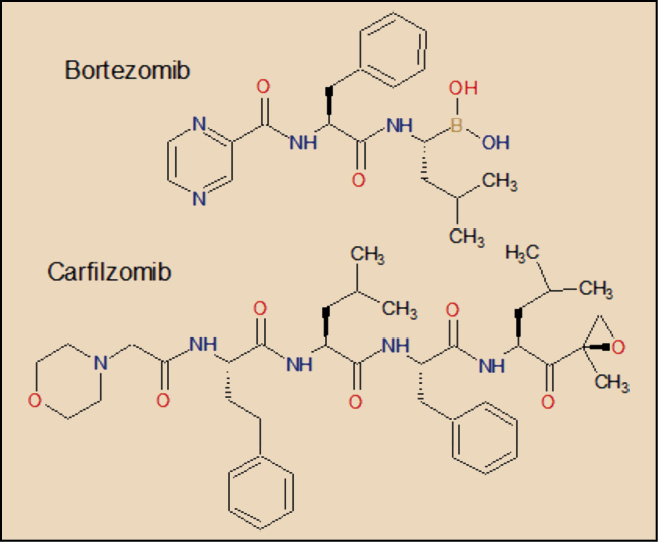
El carfilzomib es un tetrapéptido que presenta un agrupamiento de epoxicetona – altamente reactivo – en el extremo de su molécula. Se une de forma selectiva e irreversible al resto de treonina en el extremo N-terminal de los sitios activos del proteasoma 20S, el núcleo proteolítico del proteasoma 26S. Carece de actividad significativa sobre otros complejos o enzimas proteolíticos. Está estrechamente relacionado con el bortezomib, aunque carece del resto de ácido borónico característico de este último. La sustitución de este agrupamiento por la epoxicetona en el carfilzomib hace que este gane en selectividad de acción sobre el proteasoma y, al mismo tiempo, determina el carácter irreversible de su unión al mismo.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del carfilzomib han sido adecuadamente contrastadas en las indicaciones autorizadas mediante dos ensayos clínicos de fase 3 (confirmatorios de eficacia y seguridad), aleatorizados y multicéntricos.
En el primero de estos estudios (ASPIRE; Stewart, 2015) se asignó aleatoriamente a 792 pacientes con mieloma múltiple en recaída a un tratamiento a base de carfilzomib, lenalidomida y dexametasona (grupo carfilzomib) o de lenalidomida y dexametasona (grupo control), en el que la variable primaria de eficacia fue la supervivencia en ausencia de progresión tumoral (supervivencia libre de progresión). Los resultados mostraron que los pacientes del grupo carfilzomib tuvieron una mediana de supervivencia libre de progresión de 26,3 meses, frente a los 17,6 meses en el grupo control (tasa de riesgo de progresión, HR=0,69; IC95% 0,57 a 0,83; p=0,0001). Por otro lado, las tasas de supervivencia global (curvas de Kaplan-Meier) a los 24 meses fueron del 73,3 vs. 65,0%, con una reducción del riesgo de muerte del 21% para el grupo carfilzomib (HR=0,79; IC95% 0,63 a 0,99; p=0,04); asimismo, las tasas de respuesta global (completa o parcial) fueron del 87,1 vs. 66,7%, con respuestas completas en el 31,8 vs. 9,3%.
En general, los efectos secundarios más frecuentemente (<20%) asociados al carfilzomib en asociación a lenalidomida y dexametasona, fueron anemia, cansancio, diarrea, trombocitopenia, náuseas, fiebre, disnea, infecciones respiratorias, tos y edema periférico. La incidencia de efectos adversos de grado 3 o superior fue del 83,7 vs. 80,7%, obligando a suspender el tratamiento por este motivo en el 15,3 vs. 17,7%. En concreto, los efectos más graves fueron insuficiencia cardiaca (6,4% en el grupo carfilzomib vs. 4,1% en el grupo control), infarto de miocardio (3,3 vs. 1,3%), disnea (22,7 vs. 18,0%), insuficiencia renal aguda (8,4 vs. 7,2%), síndrome lítico tumoral (0,8 vs. 0%), reacciones asociadas a la infusión IV (42%), insuficiencia/fibrosis/cirrosis hepática (2,0 vs. 0,5%), neutropenia febril (3,3 vs. 1,3%).
El segundo de estos estudios (ENDEAVOR; Dimopoulos, 2016) se llevó a cabo sobre 929 pacientes con mieloma múltiple en recaída o refractario a al menos un tratamiento (hasta tres), que fueron aleatoriamente asignados a recibir de carfilzomib o bortezomib, ambos en combinación con dexametasona. Los pacientes recibieron, hasta que experimentaron progresión tumoral, carfilzomib (20 mg/m2 los días 1 y 2 del primer ciclo y 56 mg/m2 los ciclos subsiguientes, en infusión IV de 30 minutos de duración) y dexametasona (20 mg mg, oral o IV), o bortezomib (1,3 mg/m2 en bolo IV o SC) y dexametasona. Como variable primaria de eficacia se estableció la supervivencia libre de progresión.
Los resultados preliminares registrados tras un seguimiento de 11-12 meses mostraron una mediana de supervivencia libre de progresión de 18,7 meses con carfilzomib (IC95% 15,6 a no estimable) vs. 9,4 meses con bortezomib (IC95% 8,4 a 10,4), con una reducción de la tasa de progresión tumoral del 47% (HR=0,53; IC95% 0,44 a 0,65; p<0,0001). Por su parte, la tasa de respuesta global objetiva fue del 76,9 vs. 62,6% (p<0,001), siendo completa en el 12,5 vs. 6,2%. La tasa de muertes asociadas al tratamiento durante el periodo de seguimiento fue del 3,9 vs. 3,4%. La incidencia de eventos adversos graves fue del 48% con carfilzomib vs. 36% con bortezomib, siendo los más comunes (grado ≥3) anemia (14 vs. 10%), hipertensión (9 vs. 3%), trombocitopenia (8 vs. 9%) y neumonía (7 vs. 8%). Se suspendió el tratamiento por estos motivos en el 14,0 vs. 15,7% de los pacientes.
ASPECTOS INNOVADORES
El carfilzomib es un antineoplásico que actúa como un inhibidor selectivo e irreversible de la actividad quimotripsina del complejo enzimático proteasoma 20S, lo que induce la acumulación de proteínas poliubiquitinadas proaptóticas y esto, en última instancia, provoca la detención del ciclo celular, facilita la apoptosis e inhibe la tumorigénesis. Ha sido autorizado para el tratamiento, en combinación con lenalidomida y dexametasona o dexametasona sola, de pacientes adultos con mieloma múltiple que han recibido como mínimo un tratamiento previo.
Los datos clínicos disponibles apoyan claramente la eficacia del carfilzomib en el tratamiento de los cuadros recidivantes y refractarios a tratamientos previos. La combinación con lenalidomida y dexametasona prolonga en 8,7 meses la supervivencia libre de progresión conseguida con estos dos últimos fármacos, incrementando la supervivencia global a los 24 meses en 8,3 puntos porcentuales (73,3 vs. 65,0%), todo ello con tasas de respuesta global del 87,1 vs. 66,7%, que fueron completas en el 31,8 vs. 9,3%.
El otro estudio clínico enfatiza la mejora conseguida por el carfilzomib en relación a su antecesor farmacológico, el bortezomib. Aunque en este caso, no se combinó con lenalidomida, el carfilzomib incrementó la supervivencia libre de progresión en 9,3 meses (18,7 vs. 9,4) con relación al bortezomib, con una reducción de la tasa de progresión tumoral del 47% y una tasa de respuesta global objetiva del 76,9 vs. 62,6%, que fue completa en el 12,5 vs. 6,2%.
En cuanto a la seguridad, es claro que el carfilzomib es un medicamento que amplía la ya de por sí importante toxicidad de la combinación lenalidomida/dexametasona. Sin embargo, no parece que aumente el porcentaje de pacientes que se van obligados a suspender el tratamiento por este motivo (en realidad, es menor: 15,3 vs. 17,7%). En general, los efectos secundarios más frecuentes son anemia, cansancio, diarrea, trombocitopenia, náuseas, fiebre, disnea, infecciones respiratorias, tos y edema periférico. La incidencia de efectos adversos de grado 3 o superior tampoco aumenta sustancialmente (83,7 vs. 80,7%), destacando por su relevancia la insuficiencia cardiaca (6,4% vs. 4,1%), el infarto de miocardio (3,3 vs. 1,3%), la disnea (22,7 vs. 18,0%), la insuficiencia renal aguda (8,4 vs. 7,2%), las reacciones asociadas a la infusión IV (42%), la insuficiencia/fibrosis/cirrosis hepática (2,0 vs. 0,5%) y la neutropenia febril (3,3 vs. 1,3%).
El carfilzomib supone una evolución en el ámbito de los inhibidores del proteasoma, una herramienta celular fundamental en el control del ciclo vital celular y de la apoptosis, particularmente en el ámbito tumoral. Su efecto irreversible y su mayor selectividad sobe la función enzimática de tipo quimotripsina del proteasoma en relación a su antecesor, el bortezomib, se traduce en un efecto antitumoral más potente y eficaz que el de éste, pero también algo más tóxico.
A pesar de las mejoras incorporadas en el tratamiento del mieloma, especialmente con el bortezomib y la pomalidomida (tras la talidomida y lenalidomida), la mayoría de los casos acaban por hacerse resistentes y ello sin contar con el importante porcentaje de pacientes que son refractarios a un primer tratamiento. Por ello, la incorporación del carfilzomib supone un avance moderado pero real en el tratamiento del mieloma múltiple, a la espera de la incorporación de los nuevos fármacos que están siendo estudiados o incluso han sido autorizados para su uso en el mieloma múltiple, tales como el ibrutinib (un inhibidor de las tirosina cinasas de Bruton, actualmente indicado en la leucemia de células del manto), el panobinostat (un inhibidor de la histona desacetilasa), el siltuximab (inhibidor de la IL-6, actualmente autorizado en la enfermedad sistémica de Castlemann) o el daratumumab, que se une a la proteína CD38 expresada con un nivel alto en la superficie de las células tumorales del mieloma.
|
VALORACIÓN |
|
CARFILZOMIB
|
|
Grupo Terapéutico (ATC): L01XX. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES. Antineoplásicos: otros. |
|
Indicaciones autorizadas: Tratamiento, en combinación con lenalidomida y dexametasona o dexametasona sola, de pacientes adultos con mieloma múltiple que han recibido como mínimo un tratamiento previo. |
|
INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |

|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Bortezomib |
Velcade |
Janssen Cilag |
2004 |
|
Carfilzomib |
Kyprolis |
Amgen |
2016 |
BIBLIOGRAFÍA
Bibliografía
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Pomalidomida (Imnovid®) en mieloma múltiple. Panorama Actual Med. 2014; 38(375): 615-20.
- Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R, et al; ENDEAVOR Investigators. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016; 17(1): 27-38. doi: 10.1016/S1470-2045(15)00464-7.
- European Medicines Agency (EMA). Kyprolis®. European Public Assessment Report (EPAR). EMA/404277/2016; EMEA/H/C/003790. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003790/WC500197694.pdf
- Naymagon L, Abdul-Hay M. Novel agents in the treatment of multiple myeloma: a review about the future. J Hematol Oncol. 2016; 9(1): 52. doi: 10.1186/s13045-016-0282-1.
- Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al; ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015; 372(2): 142-52. doi: 10.1056/NEJMoa1411321.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
